Un contig (de contigu ) est un ensemble de segments d'ADN qui se chevauchent et qui représentent ensemble une région consensus de l'ADN . Dans les projets de séquençage ascendant , un contig fait référence à des données de séquence qui se chevauchent ( lectures ) ; dans les projets de séquençage descendant , un contig fait référence aux clones qui se chevauchent et qui forment une carte physique du génome qui est utilisée pour guider le séquençage et l'assemblage . Les contigs peuvent ainsi faire référence à la fois à des séquences d'ADN qui se chevauchent et à des segments physiques qui se chevauchent (fragments) contenus dans des clones selon le contexte.
Définition originale de contig
En 1980, Staden écrivait : « Afin de faciliter la discussion sur nos données obtenues par la méthode de séquençage par fusil de chasse, nous avons inventé le mot « contig ». Un contig est un ensemble de lectures de gel qui sont liées les unes aux autres par le chevauchement de leurs séquences. Toutes les lectures de gel appartiennent à un et un seul contig, et chaque contig contient au moins une lecture de gel. Les lectures de gel d'un contig peuvent être additionnées pour former une séquence consensus contiguë et la longueur de cette séquence est la longueur du contig. »
Contigs de séquence
Un contig de séquence est une séquence continue (non contiguë) résultant du réassemblage des petits fragments d'ADN générés par des stratégies de séquençage ascendant . Cette signification de contig est cohérente avec la définition originale de Rodger Staden (1979). de séquençage d'ADN ascendant consiste à découper l'ADN génomique en de nombreux petits fragments (« bottom »), à séquencer ces fragments, à les réassembler en contigs et finalement en génome entier (« up »). Étant donné que la technologie actuelle ne permet le séquençage direct que de fragments d'ADN relativement courts (300 à 1 000 nucléotides), l'ADN génomique doit être fragmenté en petits morceaux avant le séquençage. Dans les projets de séquençage ascendant, l'ADN amplifié est découpé de manière aléatoire en fragments de taille appropriée pour le séquençage. Les lectures de séquences suivantes, qui sont les données contenant les séquences des petits fragments, sont placées dans une base de données. Le logiciel d'assemblage recherche ensuite dans cette base de données des paires de lectures qui se chevauchent. L'assemblage des lectures d'une telle paire (comprenant, bien sûr, une seule copie de la séquence identique) produit une lecture contiguë (contig) plus longue de l'ADN séquencé. En répétant ce processus plusieurs fois, d'abord avec les paires de lectures courtes initiales, puis en utilisant des paires de plus en plus longues qui sont le résultat de l'assemblage précédent, la séquence d'ADN d'un chromosome entier peut être déterminée.
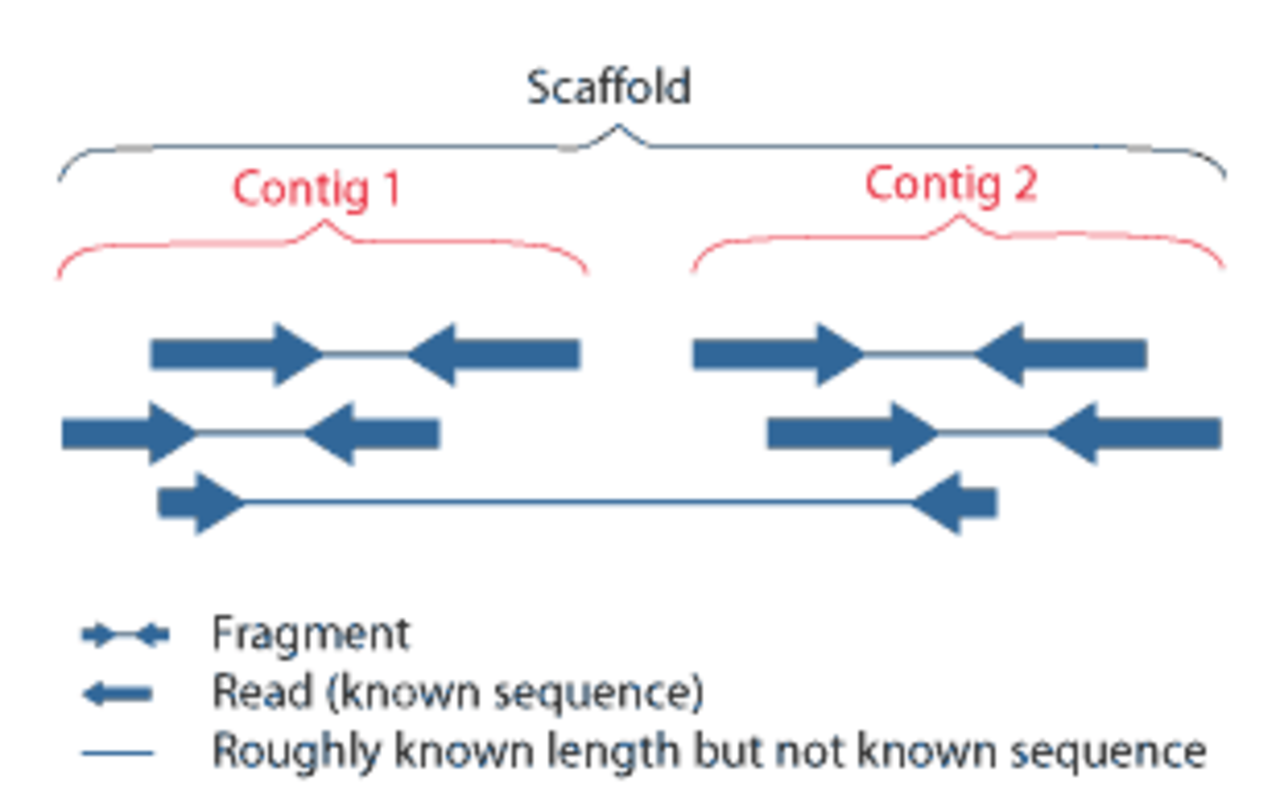
Aujourd'hui, il est courant d'utiliser la technologie de séquençage par paires , où les deux extrémités de fragments d'ADN plus longs et de taille constante sont séquencées. Ici, un contig fait toujours référence à toute séquence contiguë de données de séquence créée par chevauchement de lecture. Étant donné que les fragments ont une longueur connue, la distance entre les deux lectures d'extrémité de chaque fragment est connue. Cela donne des informations supplémentaires sur l'orientation des contigs construits à partir de ces lectures et permet leur assemblage en échafaudages dans un processus appelé échafaudage .
Les échafaudages sont constitués de contigs superposés séparés par des espaces de longueur connue. Les nouvelles contraintes imposées à l'orientation des contigs permettent le placement de séquences hautement répétées dans le génome. Si une extrémité de lecture possède une séquence répétitive, tant que sa paire de partenaires est située dans un contig, son placement est connu. Les espaces restants entre les contigs dans les échafaudages peuvent ensuite être séquencés par diverses méthodes, notamment l'amplification par PCR suivie d'un séquençage (pour les espaces plus petits) et les méthodes de clonage BAC suivies d'un séquençage pour les espaces plus grands.
Contigs BAC
Le terme contig peut également faire référence aux clones qui se chevauchent et qui forment une carte physique d'un chromosome lorsque la stratégie de séquençage descendant ou hiérarchiquecarte à faible résolution est réalisée avant le séquençage afin de fournir un cadre pour guider l'assemblage ultérieur des lectures de séquence du génome. Cette carte identifie les positions relatives et le chevauchement des clones utilisés pour le séquençage. Les ensembles de clones qui se chevauchent et qui forment un tronçon contigu d'ADN sont appelés contigs ; le nombre minimum de clones qui forment un contig qui couvre l'ensemble du chromosome constitue le chemin de mosaïque utilisé pour le séquençage. Une fois qu'un chemin de mosaïque a été sélectionné, ses BAC composants sont découpés en fragments plus petits et séquencés. Les contigs fournissent donc le cadre du séquençage hiérarchique.
L'assemblage d'une carte de contigs implique plusieurs étapes. Tout d'abord, l'ADN est découpé en morceaux plus gros (50 à 200 kb), qui sont clonés dans des BAC ou des PAC pour former une bibliothèque de BAC . Étant donné que ces clones doivent couvrir l'intégralité du génome/chromosome, il est théoriquement possible d'assembler un contig de BAC qui couvre l'intégralité du chromosome. La réalité, cependant, n'est pas toujours idéale. Des lacunes subsistent souvent, et un échafaudage, composé de contigs et de lacunes, qui couvre la région de la carte est souvent le premier résultat. Les lacunes entre les contigs peuvent être comblées par diverses méthodes décrites ci-dessous.
Construction de contigs BAC
Les contigs BAC sont construits en alignant les régions BAC dont le chevauchement est connu via une variété de méthodes. Une stratégie courante consiste à utiliser la cartographie du contenu des sites marqués par séquence (STS) pour détecter des sites d'ADN uniques en commun entre les BAC. Le degré de chevauchement est estimé approximativement par le nombre de marqueurs STS en commun entre deux clones, plus de marqueurs en commun signifie un chevauchement plus important. Étant donné que cette stratégie ne fournit qu'une estimation très approximative du chevauchement, l'analyse des fragments de digestion de restriction , qui fournit une mesure plus précise du chevauchement des clones, est souvent utilisée. Dans cette stratégie, les clones sont traités avec une ou deux enzymes de restriction et les fragments résultants sont séparés par électrophorèse sur gel . S'il s'agit de deux clones, ils auront probablement des sites de restriction en commun et partageront donc plusieurs fragments. Étant donné que le nombre de fragments en commun et la longueur de ces fragments sont connus (la longueur est jugée par comparaison à une norme de taille), le degré de chevauchement peut être déduit avec un degré élevé de précision.
Écarts entre les contigs
Des lacunes subsistent souvent après la construction initiale du contig BAC. Ces lacunes se produisent si la bibliothèque de chromosomes artificiels bactériens (BAC) examinée présente une faible complexité, ce qui signifie qu'elle ne contient pas un nombre élevé de sites STS ou de restriction, ou si certaines régions étaient moins stables dans les hôtes de clonage et donc sous-représentées dans la bibliothèque. Si des lacunes entre les contigs subsistent après que la cartographie des repères STS et l'empreinte de restriction ont été effectuées, le séquençage des extrémités des contigs peut être utilisé pour combler ces lacunes. Cette stratégie de séquençage des extrémités crée essentiellement un nouveau STS avec lequel examiner les autres contigs. Alternativement, la séquence de fin d'un contig peut être utilisée comme amorce pour traverser l'écart.