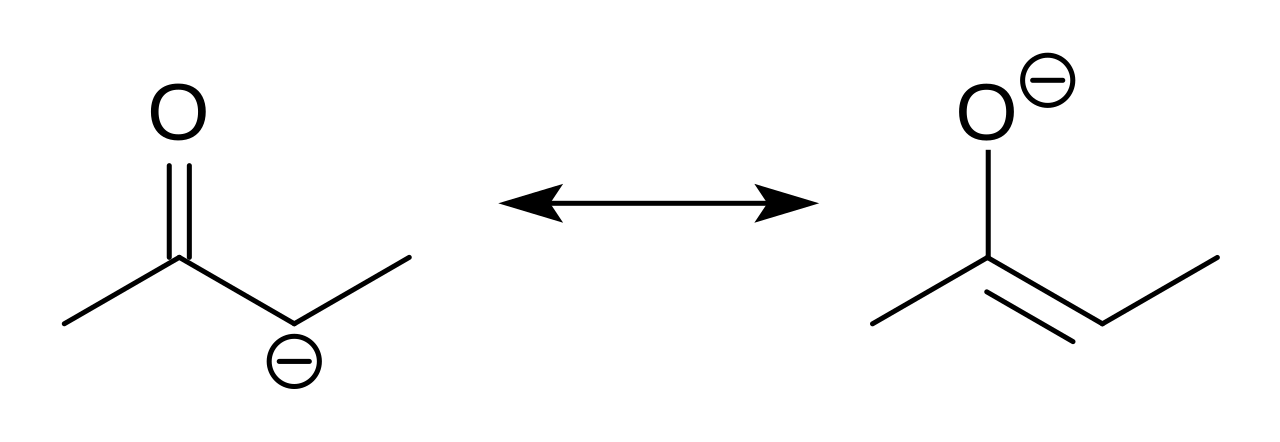
En chimie organique , les énolates sont des anions organiques issus de la déprotonation de composés carbonylés ( RR'C=O ). Rarement isolés, ils sont largement utilisés comme réactifs dans la synthèse de composés organiques .
Liaison et structure
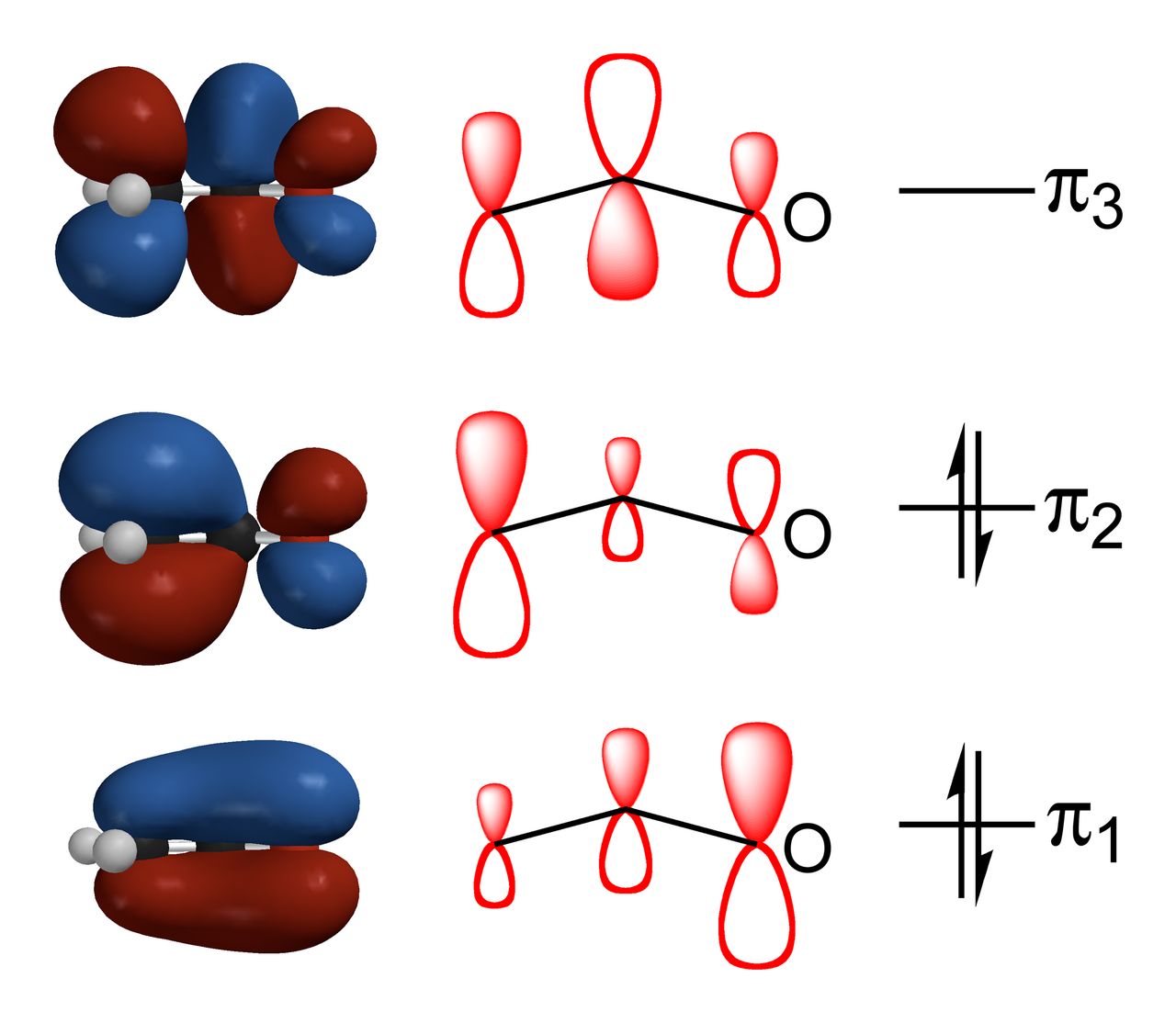
Les anions énolates sont électroniquement apparentés aux anions allyle. La charge anionique est délocalisée sur l'oxygène et les deux sites carbonés. Ils ont donc à la fois le caractère d'un alcoxyde et d'un carbanion .
Bien qu'ils soient souvent représentés comme des sels simples, ils adoptent en fait des structures complexes comportant souvent des agrégats.
=CMe2(Litmeda)_dimer_(VITLUG).png/1280px-Structure_of_PhC(OLi)=CMe2(Litmeda)_dimer_(VITLUG).png)
Préparation
La déprotonation des cétones énolisables, des alcools aromatiques, des aldéhydes et des esters donne des énolates. Avec des bases fortes, la déprotonation est quantitative. Les énolates sont généralement générés à partir de l'utilisation de diisopropylamide de lithium (LDA).
Souvent, comme dans les condensations de Claisen classiques , les réactions de Mannich et les condensations aldoliques , les énolates sont générés en faibles concentrations avec des bases alcoxydes. Dans de telles conditions, ils existent en faibles concentrations, mais ils subissent toujours des réactions avec des électrophiles. De nombreux facteurs affectent le comportement des énolates, en particulier le solvant, les additifs (par exemple les diamines) et le contre-cation (Li + contre Na + , etc.). Pour les cétones asymétriques, il existe des méthodes permettant de contrôler la régiochimie de la déprotonation.
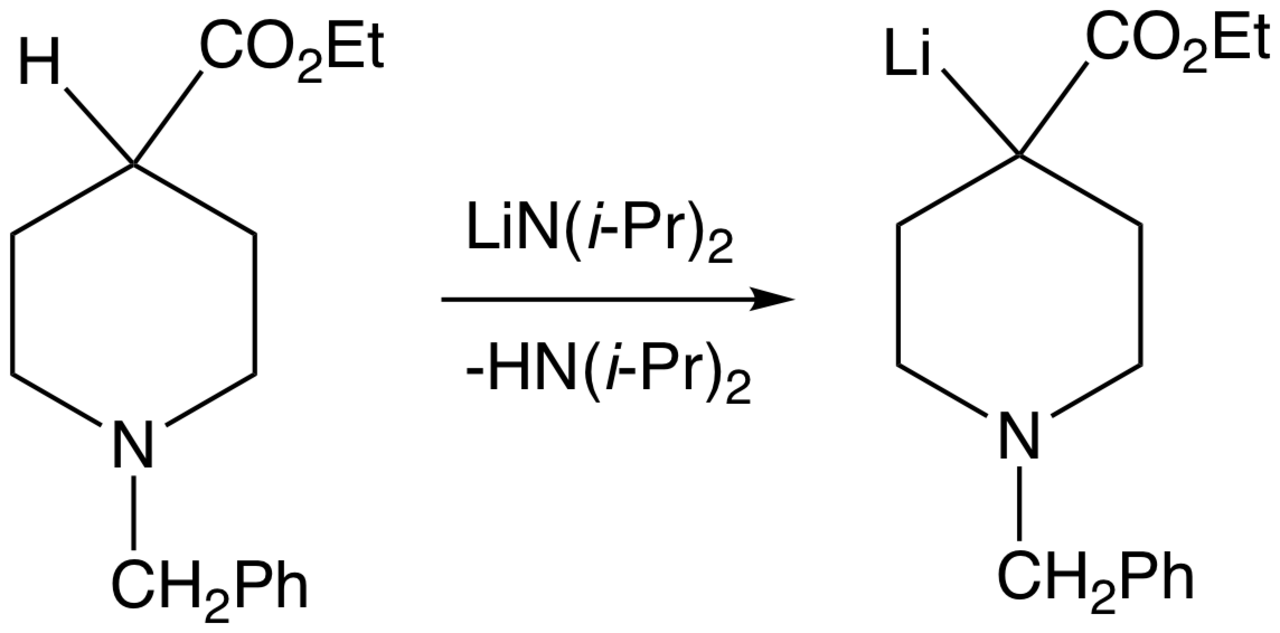
La déprotonation des acides carbonés peut se dérouler avec un contrôle de réaction cinétique ou thermodynamique . Par exemple, dans le cas de la phénylacétone , la déprotonation peut produire deux énolates différents. Il a été démontré que le LDA déprotonait le groupe méthyle, ce qui est le cours cinétique de la déprotonation. Pour assurer la production du produit cinétique, un léger excès (1,1 équivalent) de diisopropylamide de lithium est utilisé et la cétone est ajoutée à la base à −78 °C. Étant donné que la cétone est rapidement et quantitativement convertie en énolate et que la base est présente en excès à tout moment, la cétone est incapable d'agir comme navette protonique pour catalyser la formation progressive du produit thermodynamique. Une base plus faible telle qu'un alcoxyde , qui déprotone le substrat de manière réversible, permet d'obtenir l'énolate benzylique plus stable thermodynamiquement.
Les énolates peuvent être piégés par acylation et silylation , qui se produisent au niveau de l'oxygène. Les éthers d'énols silylés sont des réactifs courants en synthèse organique, comme l'illustre la réaction d'aldolisation de Mukaiyama :
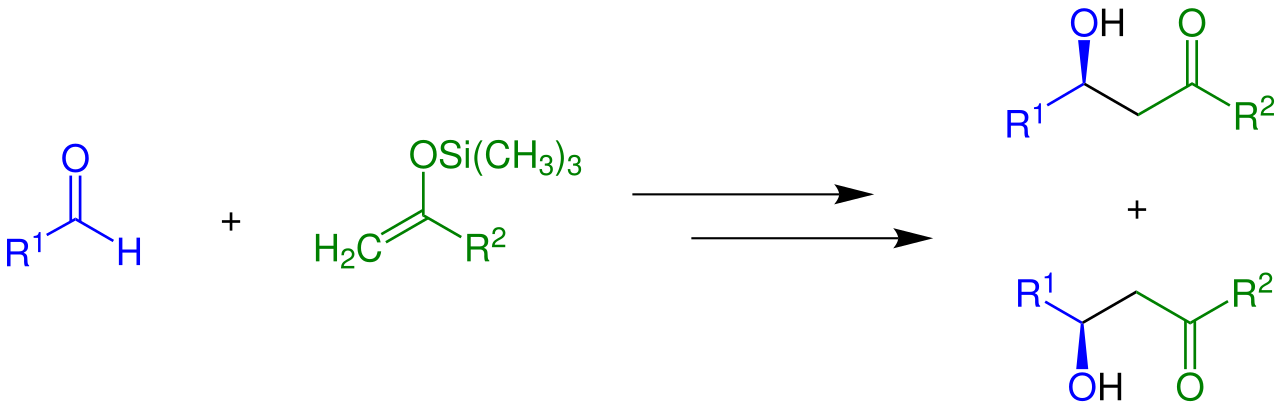
Rôle des acides de Lewis dans la formation d'énolates
En plus de l'utilisation de bases fortes, des énolates peuvent être générés en utilisant un acide de Lewis et une base faible (« conditions douces ») :

Pour que la déprotonation se produise, l'exigence stéréoélectronique est que la liaison sigma alpha-CH doit pouvoir chevaucher l'orbitale pi* du carbonyle :
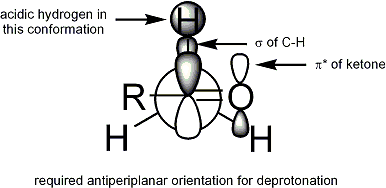
Géométrie
Des études approfondies ont été réalisées sur la formation des énolates. Il est possible de contrôler la géométrie de l'énolate :
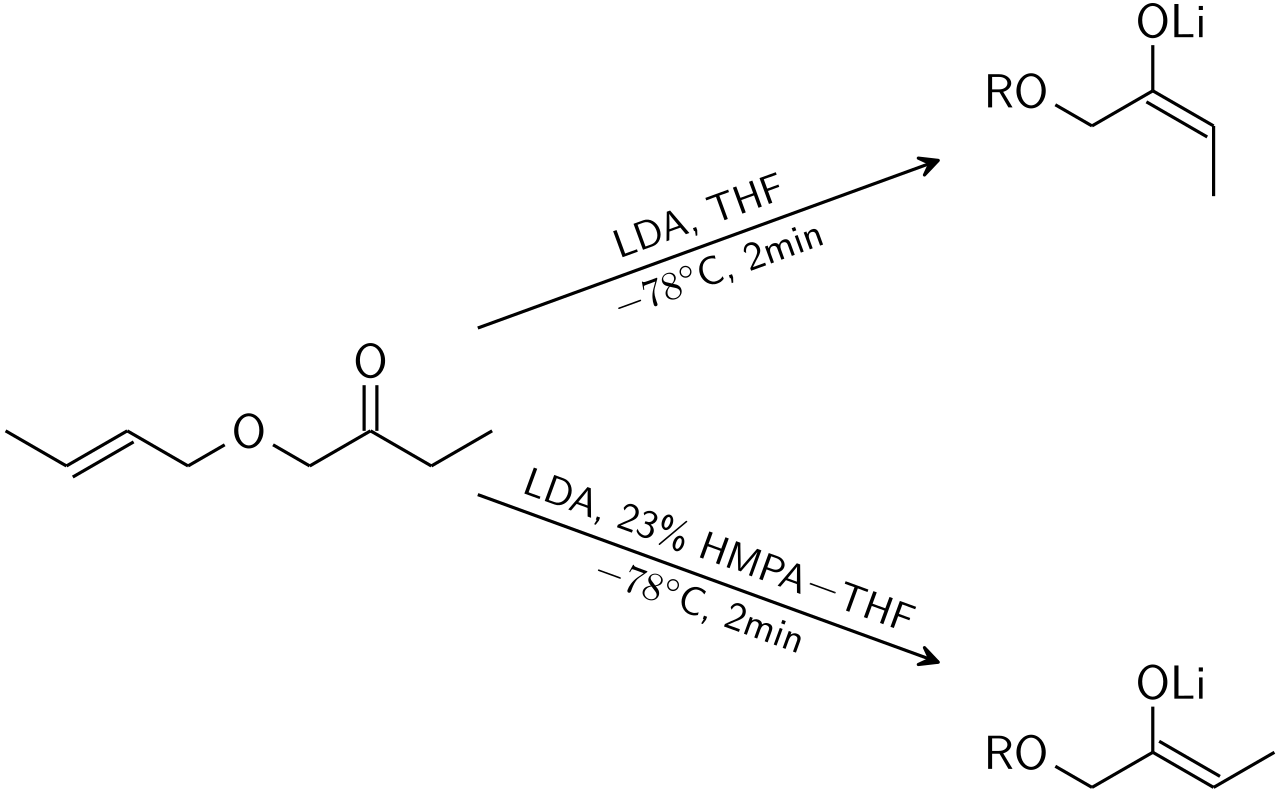
Pour les cétones, la plupart des conditions d'énolisation donnent des énolates Z. Pour les esters , la plupart des conditions d'énolisation donnent des énolates E. L'ajout de HMPA est connu pour inverser la stéréosélectivité de la déprotonation.
La formation stéréosélective des énolates a été rationalisée avec le modèle d'Irlande [ bien que sa validité soit quelque peu discutable. Dans la plupart des cas, on ne sait pas quels intermédiaires, le cas échéant, sont de nature monomérique ou oligomérique ; néanmoins, le modèle d'Irlande reste un outil utile pour comprendre les énolates.
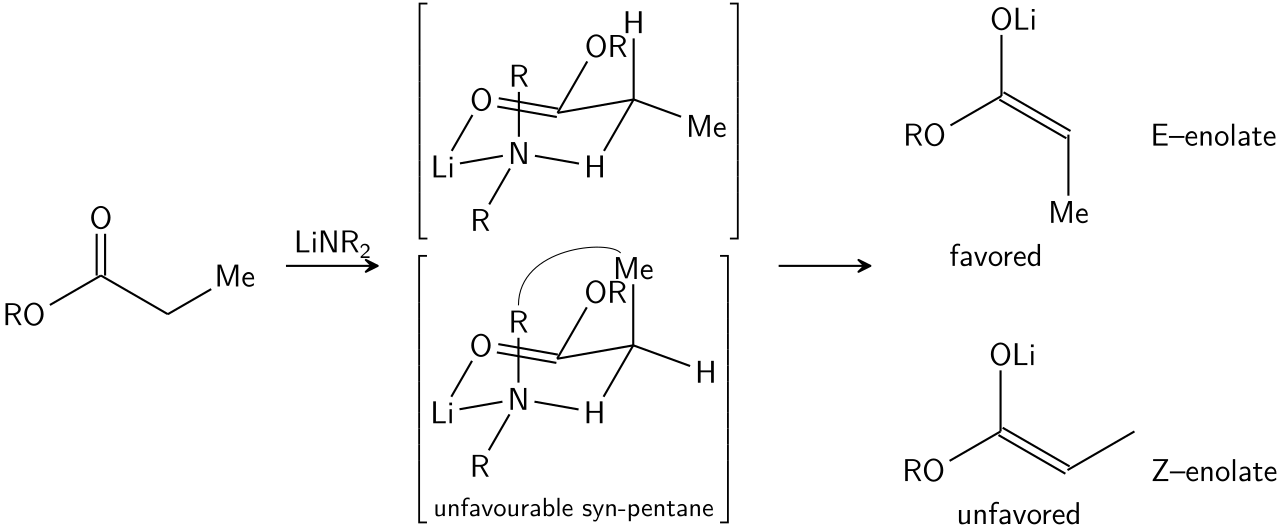
Dans le modèle d'Irlande, la déprotonation est supposée se dérouler par un état de transition monomérique à six chaînons ou cyclique . Le plus grand des deux substituants sur l'électrophile (dans le cas ci-dessus, le méthyle est plus grand que le proton) adopte une disposition équatoriale dans l'état de transition favorisé, ce qui conduit à une préférence pour les énolates E. Le modèle échoue clairement dans de nombreux cas ; par exemple, si le mélange de solvants passe du THF à 23 % de HMPA-THF (comme indiqué ci-dessus), la géométrie de l'énolate est inversée, ce qui est incompatible avec ce modèle et son état de transition cyclique.
Régiochimie de la formation d'énolates
Si une cétone asymétrique est soumise à une base, elle a le potentiel de former deux énolates régioisomères (en ignorant la géométrie de l'énolate). Par exemple :
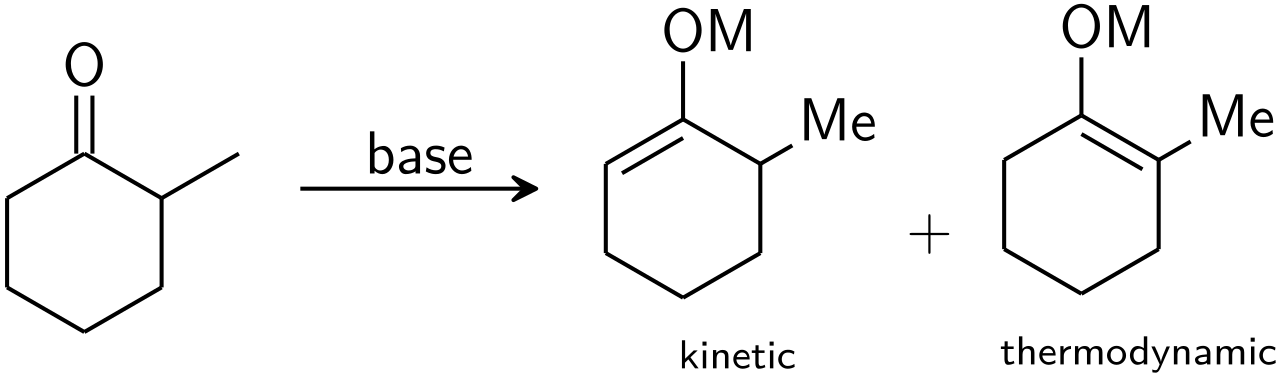
Français L'énolate trisubstitué est considéré comme l' énolate cinétique , tandis que l'énolate tétrasubstitué est considéré comme l'énolate thermodynamique. L'hydrogène alpha déprotoné pour former l'énolate cinétique est moins encombré et donc déprotoné plus rapidement. En général, les oléfines tétrasubstituées sont plus stables que les oléfines trisubstituées en raison de la stabilisation hyperconjugative. Le rapport des régioisomères d'énolate est fortement influencé par le choix de la base. Pour l'exemple ci-dessus, le contrôle cinétique peut être établi avec LDA à −78 °C, donnant une sélectivité de 99:1 de l'énolate cinétique: thermodynamique, tandis que le contrôle thermodynamique peut être établi avec du triphénylméthyllithium à température ambiante , donnant une sélectivité de 10:90.
En général, les énolates cinétiques sont favorisés par les basses températures, les conditions qui donnent une liaison métal-oxygène relativement ionique et une déprotonation rapide en utilisant un léger excès d'une base forte stériquement encombrée. La grande base ne déprotone que l'hydrogène le plus accessible, et les basses températures et l'excès de base aident à éviter l'équilibrage vers l'énolate alternatif plus stable après la formation initiale de l'énolate. Les énolates thermodynamiques sont favorisés par des temps d'équilibrage plus longs à des températures plus élevées, des conditions qui donnent une liaison métal-oxygène relativement covalente et l'utilisation d'une légère quantité sous-stoechiométrique de base forte. En utilisant une base insuffisante pour déprotoner toutes les molécules de carbonyle, les énolates et les carbonyles peuvent échanger des protons entre eux et s'équilibrer avec leur isomère le plus stable. L'utilisation de divers métaux et solvants peut permettre de contrôler la quantité de caractère ionique dans la liaison métal-oxygène.
Réactions
En tant que puissants nucléophiles, les énolates réagissent avec une variété d'électrophiles. La stéréosélectivité et la régiosélectivité sont influencées par les additifs, le solvant, les contre-ions , etc. Lorsque les électrophiles sont des halogénures d'alkyle, un problème classique se pose : O-alkylation vs C-alkylation . Le contrôle de cette sélectivité a suscité beaucoup d'attention. La charge négative des énolates est concentrée sur l'oxygène, mais ce centre est également fortement solvaté, ce qui conduit à la C-alkylation.
D’autres électrophiles importants sont les aldéhydes/cétones et les accepteurs de Michael .
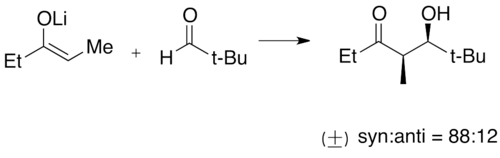
Exemple de réaction aldolique avec l'énolate de lithium
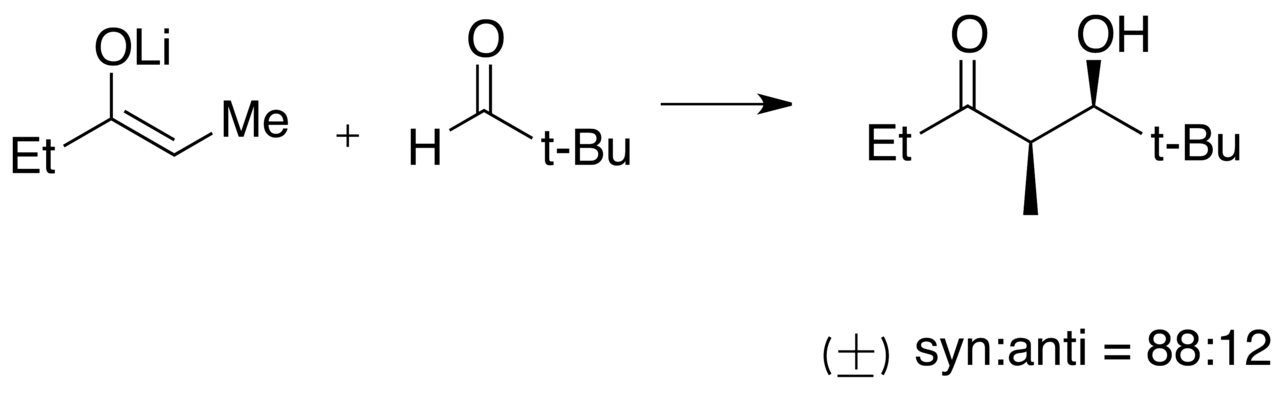
Synthèse d'énones par formation d'énolates régiospécifiques et fonctionnalité masquée


La formation régiospécifique est la formation contrôlée d'énolate par la déprotonation spécifique de l'un des carbones α de la molécule de départ de la cétone. Cela fournit l'une des stratégies de synthèse les mieux comprises pour introduire une complexité chimique dans les synthèses de produits naturels et totales . Un exemple marquant de son utilisation est la synthèse totale de la progestérone illustrée dans la figure « Formation d'énolate régiospécifique dans la synthèse totale de la progestérone ».
Lorsque les cétones sont traitées avec une base , des énolates peuvent être formés par déprotonation sur l'un ou l'autre des carbones α. La sélectivité est déterminée à la fois par les effets stériques et électroniques sur les carbones α ainsi que par la base précise utilisée (voir la figure « Fonctionnalité masquée pour la formation d'énolates régiospécifiques » pour un exemple). La formation d'énolates sera favorisée thermodynamiquement au niveau du proton le plus acide, ce qui dépend de la stabilisation électronique de l' anion résultant . Cependant, la sélectivité peut être inversée en empêchant stériquement le produit thermodynamique et donc en favorisant cinétiquement la déprotonation sur l'autre centre du carbone α. Les méthodes traditionnelles de formation d'énolates régiosélectives utilisent soit des groupes d'activation électronique (par exemple des aldéhydes ), soit des groupes de blocage stériques (par exemple une cétone protégée par un 1,2-éthanedithiol ).
Une énone peut également servir de précurseur pour la formation régiospécifique d'un énolate, l'énone étant ici une « fonctionnalité masquée » pour l'énolate. Ce processus est décrit pour la première fois par Gilbert Stork , mieux connu pour ses contributions à l'étude des méthodes de formation sélective d'énolates en synthèse organique . La réaction d'une énone avec du lithium métallique génère l'énolate au niveau du carbone α de l'énone. Le produit énolate peut être piégé ou alkylé. En utilisant la « fonctionnalité masquée », il est possible de produire des énolates qui ne sont pas accessibles par les méthodes traditionnelles.
L'approche de la « fonctionnalité masquée » pour la formation d'énolates régiospécifiques a été largement utilisée dans la synthèse totale de produits naturels. Par exemple, dans la synthèse totale de l'hormone stéroïde progestérone , Stork et ses collègues ont utilisé la « fonctionnalité masquée » pour construire de manière stéréospécifique l'un des carbones quaternaires de la molécule.
Aza énolate
Les aza énolates (également connus sous le nom d'anions imine, d'énamides, de bases de Schiff métallées et de métalloénamines) sont des analogues azotés des énolates. Lorsque les imines sont traitées avec des bases fortes telles que le LDA , des aza énolates hautement nucléophiles sont générés.
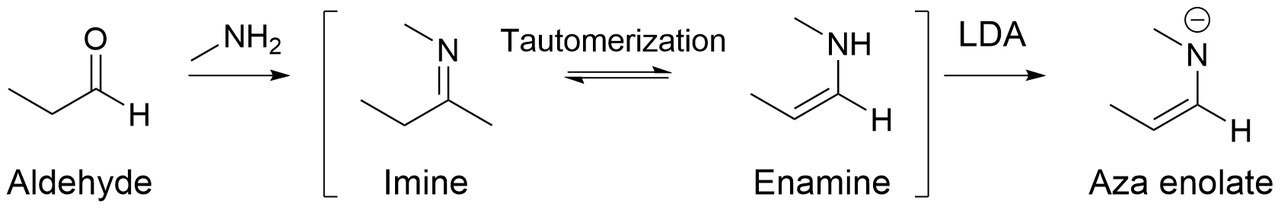
L'avantage majeur de l'utilisation d'énolates aza est qu'ils ne subissent pas d'autocondensation (c'est-à-dire une réaction d'aldolisation pour les aldéhydes ) dans une solution basique ou neutre, mais qu'ils favorisent plutôt l'alkylation sur le carbone alpha. Cela est principalement dû au fait que les imines contiennent des doubles liaisons carbone-azote contrairement aux aldéhydes, qui contiennent des doubles liaisons oxygène-carbone. Comme l'oxygène est plus électronégatif que l'azote, il retire plus de densité électronique au carbone carbonyle, induisant une charge partiellement positive plus importante sur le carbone. Par conséquent, avec plus de carbone électrophile, les aldéhydes permettent une meilleure addition nucléophile au carbone sur la double liaison carbone-oxygène.
En revanche, l'imine possède moins d'azote électronégatif , ce qui induit une charge partiellement positive plus faible sur le carbone carbonyle. Par conséquent, bien que les imines puissent toujours réagir avec les organolithiens, elles ne réagissent pas avec d'autres nucléophiles (y compris les énolates aza) pour subir des additions nucléophiles .
Au lieu de cela, les énolates d'aza réagissent de manière similaire aux énolates, formant des produits alkylés SN2 . Grâce à la conjugaison de paires non isolées d'azote, le carbone β devient un site nucléophile, permettant aux énolates d'aza de subir des réactions d'alkylation. Ainsi, les énolates d'aza peuvent réagir avec de nombreux électrophiles comme les époxydes et les halogénures d'alkyle pour former une nouvelle liaison carbone-carbone sur le carbone β.
Deux mécanismes de réaction potentiels sont présentés ci-dessous :

L'époxyde étant une molécule à trois chaînons, il présente un degré élevé de déformation du cycle . Bien que les carbones du système cyclique soient tétraédriques , préférant 109,5 degrés entre chaque atome, l'époxyde déforme les angles du cycle à 60 degrés. Pour contrer cet effet, les énolates aza nucléophiles réagissent facilement avec les époxydes pour réduire leurs déformations du cycle.
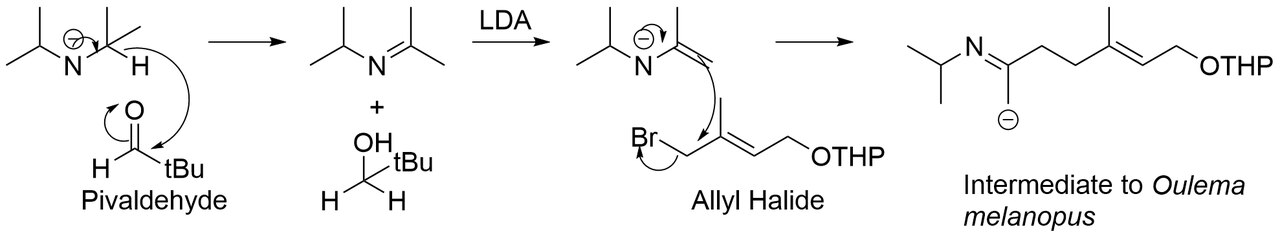
En plus de réagir avec les époxydes, les aza énolates peuvent également réagir avec les halogénures d'alkyle (ou les halogénures d'allyle comme illustré ci-dessus) pour former une nouvelle liaison sigma carbone-carbone . Cette réaction est l'une des étapes clés de la synthèse de la phéromone d'agression mâle, Oulema melanopus. L'aza énolate est généré par la réaction du LDA avec le pivaldéhyde, qui réagit ensuite avec un halogénure d'alkyle pour former un intermédiaire d'Oulema melanopus.
Les énolates d'aza peuvent également être formés avec des réactifs de Grignard et réagir avec d'autres électrophiles mous, y compris les récepteurs de Michael .