En bioinformatique , MAFFT ( alignement multiple par transformée de Fourier rapide ) est un programme permettant de créer des alignements multiples de séquences d' acides aminés ou de nucléotides . Publiée en 2002, la première version utilisait un algorithme d' alignement progressif , dans lequel les séquences étaient regroupées par transformée de Fourier rapide . Les versions ultérieures de MAFFT ont intégré d'autres algorithmes et modes de fonctionnement , notamment des options pour un alignement plus rapide d'un grand nombre de séquences , des alignements plus précis , l'alignement de séquences d'ARN non codantes .
Histoire
Il existe de nombreuses variantes du logiciel MAFFT, dont certaines sont énumérées ci-dessous :
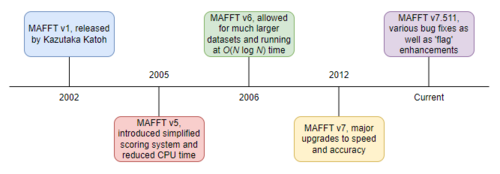
- MAFFT – La première version, créée par Kazutaka Katoh en 2002, utilisait un algorithme basé sur l'alignement progressif , dans lequel les séquences étaient regroupées à l'aide de la transformée de Fourier rapide .
- MAFFT v5 – La deuxième génération du logiciel, sortie en 2005, était une réécriture du logiciel original. Cette génération a introduit un système de score simplifié qui permet de réduire le temps de calcul et d'améliorer la précision des alignements, même pour les séquences présentant de grandes insertions ou extensions, ainsi que pour les séquences éloignées mais de longueur similaire.
- MAFFT v6 – La troisième génération, sortie en 2006, a de nouveau amélioré les versions précédentes. Elle a implémenté l'alignement de groupe à groupe, des arbres guides qui disposaient d'un algorithme de construction d'arbre approximatif mais plus rapide O ( N log N ), et a rendu la version utilisable avec des ensembles de données plus grands d'environ 50 000 séquences.
- MAFFT v7 – La quatrième génération, sortie en 2012, a considérablement amélioré la vitesse et la précision.
- MAFFT v7.511 – Une version plus récente, sortie en décembre 2022, corrige divers bogues par rapport à la version 7. Parmi les améliorations les plus notables figure une refonte de l'
--mergeoption permettant désormais le raffinement itératif , la création d'un seul algorithme MSA à partir de plusieurs sous-algorithmes MSA, ainsi que la combinaison de--mergeet--seed. Plusieurs améliorations mineures ont également été apportées à la vitesse et à la précision de MAFFT v7.
Algorithme
L'algorithme MAFFT fonctionne selon ces 5 étapes : alignement par paires, calcul de distance, construction de l'arbre guide, alignement progressif, raffinement itératif.
- Alignement par paires – Cette étape permet d'identifier les régions similaires entre les séquences d'entrée. L'algorithme commence par effectuer des alignements par paires sur l'ensemble des séquences d'entrée. La complexité temporelle de cette étape est O(L²), où L représente la longueur de la séquence.
- Matrice de distance – À partir des alignements par paires calculés, une matrice de distance est calculée afin d'évaluer la dissimilarité entre les alignements en fonction de leurs scores d'alignement . Cette étape de calcul permet de classer les séquences selon leur similarité. La complexité temporelle de la matrice de distance est de O(N²L²) , où N représente le nombre de séquences et L leur longueur. Cette complexité s'explique par le fait que le calcul de distance entre paires de séquences nécessite la comparaison de chaque position de chaque séquence.
- Arbre guide – À partir de la matrice de distance, un arbre guide est construit. Il présente une représentation hiérarchique des clusters (chaque nœud représente un cluster) et les branches indiquent la distance entre les clusters. La complexité temporelle de la construction de cet arbre guide est O(N²L) , où N est le nombre de séquences.
- Alignement progressif – L’ alignement progressif à l’aide d’un arbre guide est effectué des feuilles à la racine. L’algorithme utilise les séquences d’entrée et aligne les nœuds enfants pour calculer un alignement consensus pour le nœud parent. Cette étape est répétée jusqu’à ce que l’arbre entier soit parcouru, aboutissant à un alignement multiple final de séquences. La complexité temporelle de la méthode d’alignement progressif est de O(N²L) + O(NL²) . Ceci s’explique par le fait que le premier terme correspond au calcul de l’arbre guide mentionné précédemment, et le second à l’alignement de groupe à groupe.
- Alignement itératif – L’étape d’affinage itératif répète l’ensemble du processus en ajustant la position des insertions et des lacunes afin d’améliorer la précision de l’alignement. La complexité temporelle de l’alignement itératif dépend du nombre d’itérations. En général, elle est de O(N²L) + O(NL²) , où N est le nombre de séquences et L leur longueur.
Entrée/sortie
Formulaire Web
Saisir
Ce programme peut prendre en entrée plusieurs séquences, qui peuvent être saisies de deux manières :
Fenêtre d'entrée de séquence

L’utilisateur peut saisir directement trois séquences ou plus dans la fenêtre de saisie, dans l’un des formats suivants : GCG , FASTA , EMBL (nucléotides uniquement), GenBank , PIR , NBRF , PHYLIP ou UniProtKB/Swiss-Prot (protéines uniquement). Les séquences partiellement formatées ne sont pas acceptées ; l’ajout d’un retour à la ligne à la fin de la séquence peut faciliter son interprétation par certaines applications. Il est également conseillé d’éviter d’utiliser des données provenant de traitements de texte, car elles peuvent contenir des caractères cachés ou de contrôle.
Téléchargement du fichier de séquence
L'utilisateur peut téléverser un fichier contenant au moins trois séquences valides dans n'importe quel format mentionné ci-dessus. Les fichiers issus d'un traitement de texte peuvent donner des résultats imprévisibles en raison de la présence de caractères cachés/de contrôle ; il est donc préférable d'enregistrer les fichiers au format Unix afin d'éviter les caractères cachés Windows . Une fois le fichier téléversé, il peut être utilisé comme entrée pour l'alignement multiple de séquences.
Sortir
L'utilisateur aura la possibilité de demander que l'alignement de séquences multiples (MSA) soit généré dans l'un des deux formats disponibles :
La valeur par défaut est : Pearson/FASTA [fasta]
Paramètres
De nombreux paramètres influencent le fonctionnement de l'algorithme MAFFT. Les adapter à vos besoins est la meilleure façon d'obtenir des résultats précis et pertinents. Les paramètres les plus importants à comprendre sont : la matrice de score, la pénalité d'ouverture d'intervalle et la pénalité d'extension d'intervalle.
- Matrice de score – Les programmes de recherche de similarité de séquences protéiques tels que BLASTP, SSEARCH (UNITÉ 3.10) et FASTA utilisent des matrices de score conçues pour identifier les relations évolutives distantes (BLOSUM62 pour BLAST, BLOSUM50 pour SSEARCH et FASTA). Différentes matrices de score de similarité sont plus efficaces à différentes distances évolutives. Les matrices de score « profondes » comme BLOSUM62 et BLOSUM50 ciblent les alignements présentant une identité de 20 à 30 %, tandis que les matrices de score « superficielles » (par exemple VTML10 à VTML80) ciblent les alignements partageant une identité de 90 à 50 %, reflétant une évolution beaucoup plus faible. Dans la version originale de MAFFT, l’équation de score est présentée ci-dessous.
- Pénalité d'ouverture d'espace – Une pénalité d'espace est un score négatif attribué à un espace dans un alignement. Elle peut être constante, auquel cas un coût fixe est appliqué à l'espace, ou linéaire, auquel cas un coût fixe est appliqué à chaque symbole inséré ou supprimé. Une pénalité d'espace affine combine les deux en appliquant une pénalité constante au premier symbole d'un espace et une autre pénalité constante à chaque symbole supplémentaire inséré ou supprimé.
- Pénalité d'extension d'espace – La pénalité d'extension d'espace est un score de coût attribué à chaque symbole d'espace supplémentaire dans une région d'espacement lors de l'alignement de séquences. Elle sert à décourager la formation de longues régions d'espacement. Elle est généralement inférieure à la pénalité d'ouverture d'espace.
Précision et résultats
MAFFT est largement considéré comme l'un des outils les plus précis et polyvalents pour l'alignement multiple de séquences en bioinformatique . En effet, des études ont démontré que MAFFT offre des performances exceptionnelles par rapport à d'autres algorithmes populaires tels que Clustal W et T-Coffee , notamment pour les grands jeux de données et les séquences présentant un degré de divergence élevé . Par exemple, dans une étude comparant les performances de divers algorithmes d'alignement sur des séquences de longueur croissante, l'algorithme FFT-NS-2 de MAFFT s'est avéré le plus rapide pour toutes les tailles de séquences testées. Ceci est dû à son utilisation de la transformée de Fourier rapide ( FFT), qui permet un alignement rapide et précis, même pour des séquences très divergentes. Grâce à la FFT, l'algorithme s'exécute en O(n²) ou O(n) selon le jeu de données. MAFFT consomme moins de ressources CPU que d'autres algorithmes présentant une précision équivalente, en particulier T-Coffee, Clustal W et Needleman-Wunsch .
Les versions ultérieures de MAFFT ont ajouté d'autres algorithmes et modes de fonctionnement, notamment des options pour un alignement plus rapide d'un grand nombre de séquences, des alignements plus précis, l'alignement de séquences d'ARN non codantes, et l'ajout de nouvelles séquences aux alignements existants.
MAFFT se distingue des autres algorithmes populaires tels que Clustal W et T-Coffee par sa grande précision, sa polyvalence et son large éventail de fonctionnalités. Il offre diverses méthodes et stratégies d'alignement, notamment le raffinement itératif et les approches basées sur la cohérence, qui améliorent encore la précision et la robustesse des alignements. De ce fait, MAFFT est largement reconnu comme un outil puissant pour l'alignement de séquences multiples et est très apprécié par la communauté scientifique.