Les techniques d'analyse de microarray sont utilisées pour interpréter les données générées par des expériences sur l'ADN ( analyse de puces à ADN ), l'ARN et les microarrays de protéines , qui permettent aux chercheurs d'étudier l'état d'expression d'un grand nombre de gènes (dans de nombreux cas, le génome entier d'un organisme ) au cours d'une seule expérience. De telles expériences peuvent générer de très grandes quantités de données, permettant aux chercheurs d'évaluer l'état général d'une cellule ou d'un organisme. Des données en si grande quantité sont difficiles, voire impossibles, à analyser sans l'aide de programmes informatiques.
Introduction
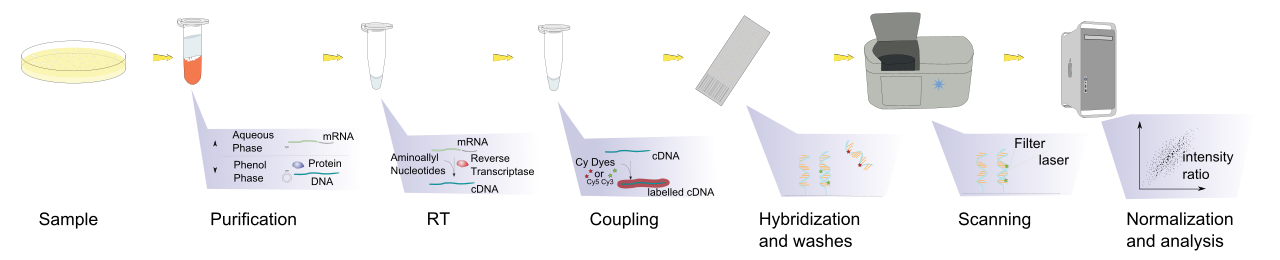
L'analyse des données de microarray est l'étape finale de la lecture et du traitement des données produites par une puce à microarray. Les échantillons subissent divers processus, notamment la purification et la numérisation à l'aide de la puce, qui produit ensuite une grande quantité de données qui nécessite un traitement via un logiciel informatique. Elle implique plusieurs étapes distinctes, comme le montre l'image ci-dessous. La modification de l'une des étapes modifiera le résultat de l'analyse, c'est pourquoi le projet MAQC .
Techniques
_(6009042166).jpg/1280px-Toxicology_Research_at_FDA_(NCTR_1470)_(6009042166).jpg)
La plupart des fabricants de microarrays, comme Affymetrix et Agilent , proposent des logiciels d'analyse de données commerciaux en plus de leurs produits de microarrays. Il existe également des options open source qui utilisent diverses méthodes pour analyser les données des microarrays.
Agrégation et normalisation
La comparaison de deux matrices différentes ou de deux échantillons différents hybridés à la même matrice implique généralement des ajustements pour les erreurs systématiques introduites par les différences de procédures et les effets d'intensité du colorant. La normalisation du colorant pour deux matrices de couleurs est souvent obtenue par régression locale . LIMMA fournit un ensemble d'outils pour la correction et la mise à l'échelle de l'arrière-plan, ainsi qu'une option pour faire la moyenne des taches dupliquées sur la lame. Une méthode courante pour évaluer le degré de normalisation d'une matrice consiste à tracer un tracé MA des données. Les tracés MA peuvent être produits à l'aide de programmes et de langages tels que R et MATLAB.
Les données brutes d'Affy contiennent environ vingt sondes pour la même cible d'ARN. La moitié d'entre elles sont des « points de discordance », qui ne correspondent pas précisément à la séquence cible. Elles peuvent théoriquement mesurer la quantité de liaison non spécifique pour une cible donnée. La moyenne multi-matrice robuste (RMA) est une approche de normalisation qui ne tire pas parti de ces points de discordance, mais doit néanmoins résumer les correspondances parfaites grâce au polissage médian . L'algorithme de polissage médian, bien que robuste, se comporte différemment selon le nombre d'échantillons analysés. La normalisation quantile , qui fait également partie de la RMA, est une approche judicieuse pour normaliser un lot de matrices afin de rendre les comparaisons ultérieures significatives.
L'algorithme actuel d'Affymetrix MAS5, qui utilise à la fois des sondes de correspondance parfaite et de non-correspondance, continue de jouir d'une grande popularité et de bien se comporter dans les tests comparatifs.
L'analyse factorielle pour la synthèse robuste des microarrays (FARMS) est une technique basée sur un modèle permettant de résumer les données des matrices au niveau de la sonde de correspondance parfaite. Elle est basée sur un modèle d'analyse factorielle pour lequel une méthode bayésienne maximale a posteriori optimise les paramètres du modèle sous l'hypothèse d'un bruit de mesure gaussien. Selon le benchmark Affycomp FARMS a surpassé toutes les autres méthodes de synthèse en termes de sensibilité et de spécificité.
Identification d'une expression différentielle significative
Il existe de nombreuses stratégies pour identifier les sondes de matrice qui présentent un niveau inhabituel de surexpression ou de sous-expression. La plus simple consiste à qualifier de « significative » toute sonde qui diffère en moyenne d'au moins deux fois entre les groupes de traitement. Des approches plus sophistiquées sont souvent liées à des tests t ou à d'autres mécanismes qui prennent en compte à la fois la taille de l'effet et la variabilité. Curieusement, les valeurs p associées à des gènes particuliers ne se reproduisent pas bien entre les expériences répétées, et les listes générées par un changement de pli direct fonctionnent beaucoup mieux. Cela représente une observation extrêmement importante, car le but de la réalisation d'expériences est de prédire le comportement général. Le groupe MAQC recommande d'utiliser une évaluation du changement de pli plus une valeur p non stricte, soulignant en outre que les changements dans la correction de fond et le processus de mise à l'échelle n'ont qu'un impact minimal sur l'ordre de classement des différences de changement de pli, mais un impact substantiel sur les valeurs p.
Regroupement
Le clustering est une technique d'exploration de données utilisée pour regrouper des gènes ayant des profils d'expression similaires. Le clustering hiérarchique et le clustering k-means sont des techniques largement utilisées dans l'analyse des microarrays.
Regroupement hiérarchique
Le clustering hiérarchique est une méthode statistique permettant de trouver des clusters relativement homogènes . Le clustering hiérarchique se compose de deux phases distinctes. Au départ, une matrice de distance contenant toutes les distances par paires entre les gènes est calculée. La corrélation de Pearson et la corrélation de Spearman sont souvent utilisées comme estimations de dissimilarité, mais d'autres méthodes, comme la distance de Manhattan ou la distance euclidienne , peuvent également être appliquées. Étant donné le nombre de mesures de distance disponibles et leur influence sur les résultats de l'algorithme de clustering, plusieurs études ont comparé et évalué différentes mesures de distance pour le clustering de données de microarray, en tenant compte de leurs propriétés intrinsèques et de leur robustesse au bruit. Après le calcul de la matrice de distance initiale, l'algorithme de clustering hiérarchique (A) joint de manière itérative les deux clusters les plus proches à partir de points de données uniques (approche agglomérative, ascendante, qui est assez couramment utilisée), ou (B) partitionne les clusters de manière itérative à partir de l'ensemble complet (approche descendante, par division). Après chaque étape, une nouvelle matrice de distance entre les clusters nouvellement formés et les autres clusters est recalculée. Les méthodes d'analyse de cluster hiérarchique comprennent :
- Lien simple (méthode du minimum, voisin le plus proche)
- Liaison moyenne ( UPGMA )
- Liaison complète (méthode du maximum, voisin le plus éloigné)
Différentes études ont déjà montré empiriquement que l'algorithme de clustering à liaison unique produit de mauvais résultats lorsqu'il est utilisé pour les données de puces à ADN d'expression génétique et doit donc être évité.
Regroupement K-means
Le clustering K-means est un algorithme permettant de regrouper des gènes ou des échantillons en fonction d'un motif en groupes K. Le regroupement est effectué en minimisant la somme des carrés des distances entre les données et le centroïde du cluster correspondant . Ainsi, le but du clustering K-means est de classer les données en fonction d'une expression similaire. L'algorithme de clustering K-means et certaines de ses variantes (y compris les k-médoïdes ) se sont avérés produire de bons résultats pour les données d'expression génétique (au moins meilleurs que les méthodes de clustering hiérarchique). Des comparaisons empiriques des k-means , des k-médoïdes , des méthodes hiérarchiques et de différentes mesures de distance peuvent être trouvées dans la littérature.
Reconnaissance des formes
Les systèmes commerciaux d'analyse de réseaux génétiques tels qu'Ingenuity et Pathway studio créent des représentations visuelles de gènes exprimés de manière différentielle en se basant sur la littérature scientifique actuelle. Des outils non commerciaux tels que FunRich, GenMAPP et Moksiskaan aident également à organiser et à visualiser les données de réseaux génétiques obtenues à partir d'une ou plusieurs expériences de microarray. Une grande variété d'outils d'analyse de microarray sont disponibles via Bioconductor écrit dans le langage de programmation R. Le module SAM fréquemment cité et d'autres outils de microarray sont disponibles via l'Université de Stanford. Un autre ensemble est disponible auprès de Harvard et du MIT.
Des outils logiciels spécialisés pour l'analyse statistique afin de déterminer l'étendue de la surexpression ou de la sous-expression d'un gène dans une expérience de microarray par rapport à un état de référence ont également été développés pour aider à identifier les gènes ou les ensembles de gènes associés à des phénotypes particuliers . Une de ces méthodes d'analyse, connue sous le nom d'analyse d'enrichissement d'ensemble de gènes (GSEA), utilise une statistique de style Kolmogorov-Smirnov pour identifier les groupes de gènes qui sont régulés ensemble. Ce progiciel de statistiques tiers offre à l'utilisateur des informations sur les gènes ou les ensembles de gènes d'intérêt, y compris des liens vers des entrées dans des bases de données telles que GenBank du NCBI et des bases de données organisées telles que Biocarta et Gene Ontology . L'outil d'analyse d'enrichissement de complexes protéiques (COMPLEAT) fournit une analyse d'enrichissement similaire au niveau des complexes protéiques. L'outil peut identifier la régulation dynamique du complexe protéique dans différentes conditions ou à différents moments. Des systèmes connexes, PAINT et SCOPE aux facteurs de transcription précédemment identifiés . Un autre outil d'analyse statistique est Rank Sum Statistics for Gene Set Collections (RssGsc), qui utilise des fonctions de distribution de probabilité de somme de rang pour trouver des ensembles de gènes qui expliquent les données expérimentales. Une autre approche est la méta-analyse contextuelle, c'est-à-dire la découverte de la façon dont un groupe de gènes réagit à une variété de contextes expérimentaux. Genevestigator est un outil public permettant d'effectuer une méta-analyse contextuelle dans des contextes tels que les parties anatomiques, les stades de développement et la réponse aux maladies, aux produits chimiques, aux stress et aux néoplasmes .
Analyse de la signification des microarrays (SAM)
L'analyse de la signification des microarrays (SAM) est une technique statistique , établie en 2001 par Virginia Tusher, Robert Tibshirani et Gilbert Chu , pour déterminer si les changements dans l'expression des gènes sont statistiquement significatifs. Avec l'avènement des microarrays d'ADN , il est désormais possible de mesurer l'expression de milliers de gènes dans une seule expérience d'hybridation. Les données générées sont considérables, et une méthode permettant de trier ce qui est significatif et ce qui ne l'est pas est essentielle. SAM est distribué par l'Université de Stanford dans un package R. ]
La SAM identifie les gènes statistiquement significatifs en effectuant des tests t spécifiques aux gènes et calcule une statistique d j pour chaque gène j , qui mesure la force de la relation entre l'expression des gènes et une variable de réponse. Cette analyse utilise des statistiques non paramétriques , car les données peuvent ne pas suivre une distribution normale . La variable de réponse décrit et regroupe les données en fonction des conditions expérimentales. Dans cette méthode, des permutations répétées des données sont utilisées pour déterminer si l'expression d'un gène est significativement liée à la réponse. L'utilisation d'une analyse basée sur la permutation tient compte des corrélations dans les gènes et évite les hypothèses paramétriques sur la distribution des gènes individuels. Il s'agit d'un avantage par rapport à d'autres techniques (par exemple, ANOVA et Bonferroni ), qui supposent une variance égale et/ou une indépendance des gènes.
Protocole de base
- Réaliser des expériences de microréseaux : microréseaux d’ADN avec amorces oligo et ADNc, réseaux SNP, réseaux de protéines, etc.
- Analyse des expressions d'entrée dans Microsoft Excel — voir ci-dessous
- Exécutez SAM en tant que module complémentaire Microsoft Excel
- Ajustez le paramètre de réglage Delta pour obtenir un nombre significatif de gènes ainsi qu'un taux de fausses découvertes (FDR) acceptable et évaluez la taille de l'échantillon en calculant la différence moyenne d'expression dans le contrôleur de tracé SAM
- Liste des gènes exprimés différemment (gènes exprimés positivement et négativement)
Exécution de SAM
- SAM est disponible en téléchargement en ligne sur http://www-stat.stanford.edu/~tibs/SAM/ pour les utilisateurs universitaires et non universitaires après avoir terminé une étape d'inscription.
- SAM est exécuté en tant que module complémentaire Excel et le contrôleur de tracé SAM permet la personnalisation du taux de fausses découvertes et du delta, tandis que les fonctionnalités de tracé SAM et de sortie SAM génèrent une liste de gènes significatifs, un tableau Delta et une évaluation des tailles d'échantillon.
- Les permutations sont calculées en fonction du nombre d'échantillons
- Permutations de blocs
- Les blocs sont des lots de microarrays ; par exemple, pour huit échantillons divisés en deux groupes (témoin et affecté), il y a 4!=24 permutations pour chaque bloc et le nombre total de permutations est (24)(24)= 576. Un minimum de 1000 permutations est recommandé ;
le nombre de permutations est défini par l'utilisateur lors de l'imputation des valeurs correctes pour l'ensemble de données pour exécuter SAM
Formats de réponse
Types :
- Quantitatif — valeur réelle (comme la fréquence cardiaque)
- Une classe — teste si l'expression moyenne des gènes diffère de zéro
- Deux classes — deux séries de mesures
- Non apparié — les unités de mesure sont différentes dans les deux groupes ; par exemple, groupes de contrôle et de traitement avec des échantillons de patients différents
- Appariés — les mêmes unités expérimentales sont mesurées dans les deux groupes ; par exemple, des échantillons avant et après le traitement provenant des mêmes patients
- Multiclasse — plus de deux groupes contenant chacun des unités expérimentales différentes ; généralisation de deux types de classes non appariées
- Survie — données sur le temps écoulé jusqu'à un événement (par exemple, décès ou rechute)
- Évolution dans le temps — chaque unité expérimentale est mesurée à plusieurs moments ; les unités expérimentales appartiennent à une conception à une ou deux classes
- Découverte de modèle — aucun paramètre de réponse explicite n'est spécifié ; l'utilisateur spécifie le gène propre (composant principal) des données d'expression et le traite comme une réponse quantitative
Algorithme
SAM calcule une statistique de test pour la différence relative dans l'expression des gènes en se basant sur l'analyse de permutation des données d'expression et calcule un taux de fausses découvertes. Les principaux calculs du programme sont illustrés ci-dessous.
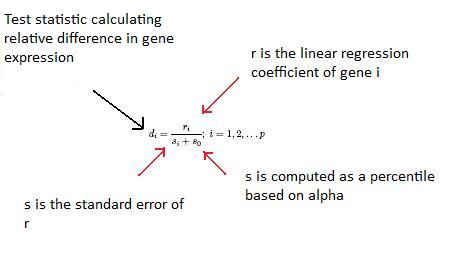

La constante s o est choisie pour minimiser le coefficient de variation de d i . r i est égal aux niveaux d'expression (x) pour le gène i dans des conditions expérimentales y.
Les variations de repli (t) sont spécifiées pour garantir que les gènes appelés changements significatifs présentent au moins une quantité prédéfinie. Cela signifie que la valeur absolue des niveaux d'expression moyens d'un gène dans chacune des deux conditions doit être supérieure à la variation de repli (t) pour être qualifiée de positive et inférieure à l'inverse de la variation de repli (t) pour être qualifiée de négative.
L'algorithme SAM peut être énoncé comme suit :
- Classement des statistiques de test en fonction de l'ampleur
- Pour chaque permutation, calculez les scores nuls ordonnés (non affectés)
- Tracez la statistique du test ordonné par rapport aux scores nuls attendus
- Chaque gène est considéré comme significatif si la valeur absolue de la statistique de test pour ce gène moins la statistique de test moyenne pour ce gène est supérieure à un seuil indiqué
- Estimez le taux de fausses découvertes en fonction des valeurs attendues par rapport aux valeurs observées
Sortir
- Ensembles de gènes importants
- Ensemble de gènes positif — une expression plus élevée de la plupart des gènes de l'ensemble de gènes est corrélée à des valeurs plus élevées du phénotype y
- Ensemble de gènes négatif — une expression plus faible de la plupart des gènes de l'ensemble de gènes est corrélée à des valeurs plus élevées du phénotype y
Fonctionnalités SAM
- Les données provenant de matrices d'oligos ou d'ADNc, de matrices de SNP, de matrices de protéines, etc. peuvent être utilisées dans SAM
- Corréler les données d’expression aux paramètres cliniques
- Corrélation des données d’expression avec le temps
- Utilise la permutation des données pour estimer le taux de fausses découvertes pour plusieurs tests
- Indique le taux de fausses découvertes locales (le FDR pour les gènes ayant un d i similaire à celui de ce gène) et les taux d'échec
- Peut fonctionner avec une conception bloquée lorsque les traitements sont appliqués dans différents lots de matrices
- Peut ajuster le seuil déterminant le nombre de gènes dits significatifs
Correction des erreurs et contrôle qualité
Contrôle de qualité
Des matrices entières peuvent présenter des défauts évidents détectables par inspection visuelle, comparaisons par paires avec des matrices du même groupe expérimental ou analyse de la dégradation de l'ARN. Les résultats peuvent être améliorés en supprimant entièrement ces matrices de l'analyse.
Correction de l'arrière-plan
Selon le type de réseau, le signal lié à la liaison non spécifique du fluorophore peut être soustrait pour obtenir de meilleurs résultats. Une approche consiste à soustraire l'intensité moyenne du signal de la zone entre les spots. Une variété d'outils de correction de l'arrière-plan et d'analyses plus approfondies sont disponibles auprès de TIGR, Agilent (GeneSpring), et Ocimum Bio Solutions (Genowiz).
Filtrage ponctuel
L'identification visuelle d'artefacts locaux, tels que des défauts d'impression ou de lavage, peut également suggérer l'élimination de taches individuelles. Cette opération peut prendre un temps considérable en fonction de la qualité de fabrication du réseau. De plus, certaines procédures nécessitent l'élimination de toutes les taches dont la valeur d'expression est inférieure à un certain seuil d'intensité.