La pathogénomique est un domaine qui utilise une technologie de criblage à haut débit et la bioinformatique pour étudier la résistance codée des microbes, ainsi que les facteurs de virulence (VF), qui permettent à un micro-organisme d'infecter un hôte et éventuellement de provoquer une maladie. Cela comprend l'étude des génomes d' agents pathogènes qui ne peuvent pas être cultivés en dehors d'un hôte. Dans le passé, les chercheurs et les professionnels de la santé trouvaient difficile d'étudier et de comprendre les caractéristiques pathogènes des organismes infectieux. Grâce à une technologie plus récente, les génomes pathogènes peuvent être identifiés et séquencés dans un délai beaucoup plus court et à moindre coût, améliorant ainsi la capacité à diagnostiquer, traiter et même prédire et prévenir les infections et les maladies pathogènes. Elle a également permis aux chercheurs de mieux comprendre les événements d'évolution du génome - perte, gain, duplication, réarrangement de gènes - et comment ces événements ont un impact sur la résistance des agents pathogènes et leur capacité à provoquer des maladies. Cet afflux d’informations a créé un besoin d’outils bioinformatiques et de bases de données pour analyser et rendre les vastes quantités de données accessibles aux chercheurs, et a soulevé des questions éthiques sur la sagesse de reconstituer des agents pathogènes auparavant éteints et mortels afin de mieux comprendre la virulence.
Histoire
Au début de l'étude de la génomique, les scientifiques trouvaient difficile de séquencer les informations génétiques. Le domaine a commencé à exploser en 1977 lorsque Fred Sanger , Ph. D., et ses collègues ont séquencé le génome basé sur l'ADN d'un bactériophage , en utilisant une méthode désormais connue sous le nom de méthode Sanger . La méthode Sanger pour le séquençage de l'ADN a fait progresser de manière exponentielle la biologie moléculaire et a directement conduit à la capacité de séquencer les génomes d'autres organismes, y compris le génome humain complet.
Le génome d'Haemophilus influenza a été l'un des premiers génomes d'organismes séquencés en 1995 par J. Craig Venter et Hamilton Smith à l'aide du séquençage shotgun du génome entier. Depuis lors, des séquençages à haut débit plus récents et plus efficaces, tels que le séquençage génomique de nouvelle génération (NGS) et le séquençage génomique à cellule unique, ont été développés. Alors que la méthode Sanger est capable de séquencer un fragment d'ADN à la fois, la technologie NGS peut séquencer des milliers de séquences à la fois. Avec la capacité de séquencer rapidement l'ADN, de nouvelles connaissances ont été développées, comme la découverte que, puisque les génomes procaryotes sont plus diversifiés qu'on ne le pensait à l'origine, il est nécessaire de séquencer plusieurs souches d'une espèce plutôt que quelques-unes seulement. E.coli est un exemple de l'importance de cette démarche, les gènes codant pour les facteurs de virulence dans deux souches de l'espèce différant d'au moins trente pour cent. Ces connaissances, ainsi qu’une étude plus approfondie du gain, de la perte et du changement du génome, donnent aux chercheurs des informations précieuses sur la manière dont les agents pathogènes interagissent dans les environnements hôtes et sur la manière dont ils sont capables d’infecter les hôtes et de provoquer des maladies.
Bioinformatique des pathogènes
Français Avec cet afflux important de nouvelles informations, il y a eu une demande accrue en bioinformatique pour que les scientifiques puissent analyser correctement les nouvelles données. En réponse, des logiciels et d'autres outils ont été développés à cette fin. De plus, depuis 2008, la quantité de séquences stockées doublait tous les 18 mois, ce qui rend urgent le besoin de meilleurs moyens d'organiser les données et d'aider la recherche. En réponse, de nombreuses bases de données accessibles au public et d'autres ressources ont été créées, notamment le programme de détection des agents pathogènes du NCBI, le Pathosystems Resource Integration Centre (PATRIC), Pathogenwatch, la Virulence Factor Database (VFDB) des bactéries pathogènes, la base de données Victors des facteurs de virulence des agents pathogènes humains et animaux. Jusqu'en 2022, les agents pathogènes les plus séquencés sont Salmonella enterica et E. coli - Shigella. Les technologies de séquençage, les outils bioinformatiques, les bases de données, les statistiques liées aux génomes pathogènes et les applications en criminalistique, en épidémiologie, en pratique clinique et en sécurité alimentaire ont été largement examinés.
Analyse microbienne
Les agents pathogènes peuvent être des procaryotes ( archées ou bactéries ), des eucaryotes unicellulaires ou des virus . Les génomes procaryotes ont généralement été plus faciles à séquencer en raison de leur taille plus petite par rapport aux eucaryotes. De ce fait, il existe un biais dans la déclaration du comportement bactérien pathogène . Indépendamment de ce biais dans la déclaration, de nombreux événements génomiques dynamiques sont similaires dans tous les types d'organismes pathogènes. L'évolution génomique se produit via le gain de gènes, la perte de gènes et le réarrangement du génome, et ces « événements » sont observés dans plusieurs génomes pathogènes, certains agents pathogènes bactériens subissant les trois. Cependant, la pathogénomique ne se concentre pas exclusivement sur la compréhension des interactions agent pathogène-hôte . La compréhension du comportement individuel ou coopératif des agents pathogènes fournit des connaissances sur le développement ou l'hérédité des facteurs de virulence des agents pathogènes. Grâce à une compréhension plus approfondie des petites sous-unités qui causent l'infection, il pourrait être possible de développer de nouvelles thérapies efficaces et rentables.
Cause et analyse de la diversité génomique
Des génomes dynamiques à forte plasticité sont nécessaires pour permettre aux agents pathogènes, en particulier aux bactéries, de survivre dans des environnements changeants. Avec l'aide de méthodes de séquençage à haut débit et de technologies in silico , il est possible de détecter, de comparer et de cataloguer bon nombre de ces événements génomiques dynamiques. La diversité génomique est importante lors de la détection et du traitement d'un agent pathogène, car ces événements peuvent modifier la fonction et la structure de l'agent pathogène. Il est nécessaire d'analyser plus d'une séquence génomique d'une espèce pathogène pour comprendre les mécanismes pathogènes. La génomique comparative est une méthodologie qui permet aux scientifiques de comparer les génomes de différentes espèces et souches. Il existe plusieurs exemples d'études génomiques comparatives réussies, parmi lesquelles l'analyse de Listeria et d'Escherichia coli . Certaines études ont tenté d'aborder la différence entre les microbes pathogènes et non pathogènes . Cette enquête s’avère toutefois difficile, car une seule espèce bactérienne peut avoir de nombreuses souches et le contenu génomique de chacune de ces souches varie.
Dynamique évolutive
Les différentes souches microbiennes et le contenu génomique sont causés par différentes forces, notamment trois événements évolutifs spécifiques qui ont un impact sur la résistance des agents pathogènes et leur capacité à provoquer des maladies : le gain de gènes, la perte de gènes et le réarrangement du génome.
Perte de gènes et dégradation du génome
La perte de gènes se produit lorsque des gènes sont supprimés. La raison pour laquelle cela se produit n'est pas encore entièrement comprise, bien qu'il s'agisse très probablement d'une adaptation à un nouvel environnement ou à une nouvelle niche écologique. Certains chercheurs pensent que la perte de gènes peut en fait augmenter la forme physique et la survie des agents pathogènes. Dans un nouvel environnement, certains gènes peuvent devenir inutiles pour la survie, et donc des mutations sont finalement « autorisées » sur ces gènes jusqu'à ce qu'ils deviennent des « pseudogènes » inactifs. Ces pseudogènes sont observés dans des organismes tels que Shigella flexneri , Salmonella enterica , et Yersinia pestis . Au fil du temps, les pseudogènes sont supprimés et les organismes deviennent totalement dépendants de leur hôte en tant qu'endosymbiotes ou agents pathogènes intracellulaires obligatoires , comme on le voit chez Buchnera , Myobacterium leprae et Chlamydia trachomatis . Ces gènes supprimés sont également appelés gènes anti-virulence (AVG) car on pense qu'ils auraient pu empêcher l'organisme de devenir pathogène. Pour être plus virulent, infecter un hôte et rester en vie, l'agent pathogène devait se débarrasser de ces AVG. Le processus inverse peut également se produire, comme cela a été observé lors de l'analyse des souches de Listeria , qui a montré qu'une taille de génome réduite conduisait à une souche de Listeria non pathogène à partir d'une souche pathogène. Des systèmes ont été développés pour détecter ces pseudogènes/AVG dans une séquence génomique.
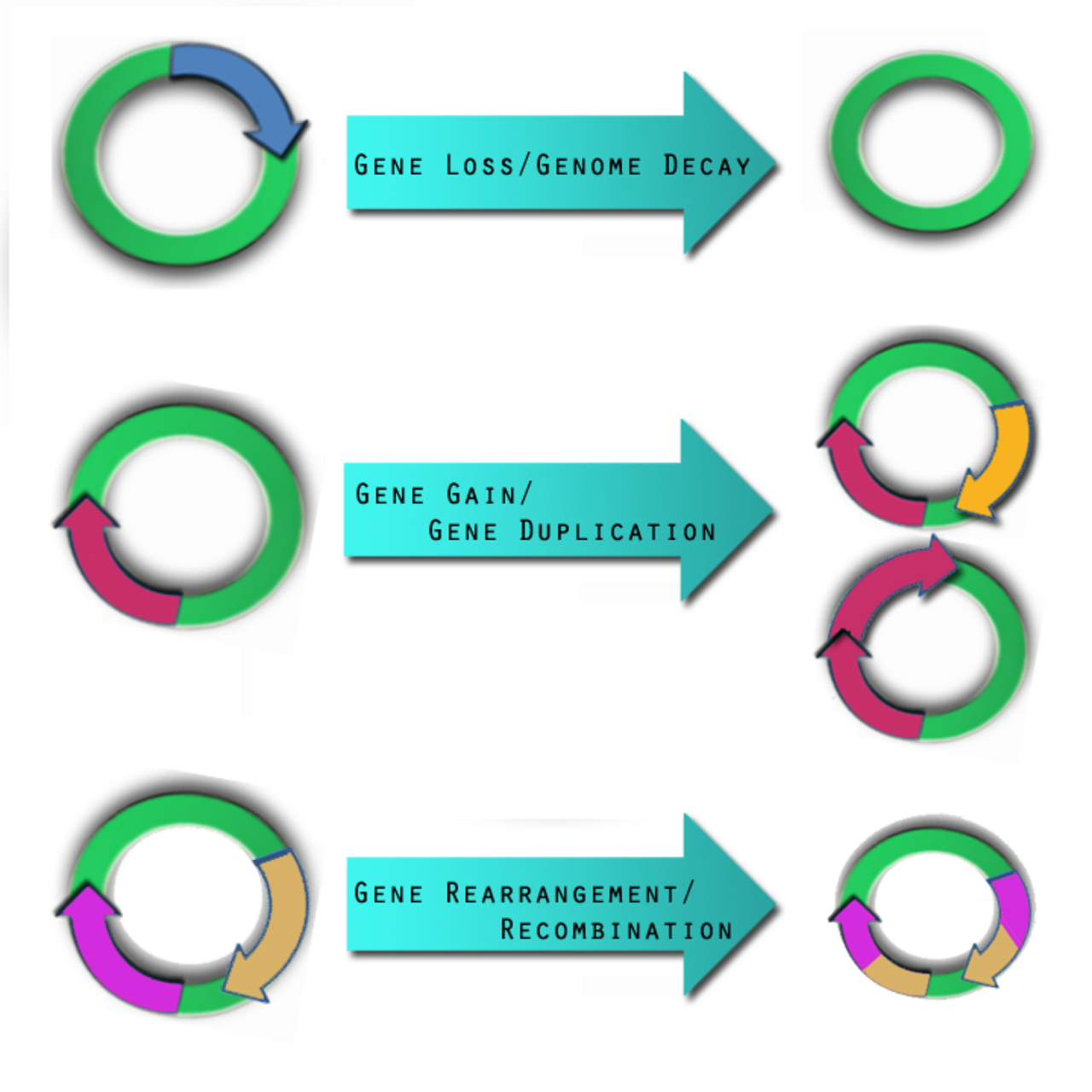
Gain et duplication de gènes
L'une des principales forces à l'origine du gain génétique est le transfert horizontal (latéral) de gènes (LGT). Il est particulièrement intéressant dans les études microbiennes car ces éléments génétiques mobiles peuvent introduire des facteurs de virulence dans un nouveau génome. Une étude comparative menée par Gill et al. en 2005 a postulé que le LGT pourrait avoir été la cause des variations pathogènes entre Staphylococcus epidermidis et Staphylococcus aureus . Il subsiste cependant un certain scepticisme quant à la fréquence du LGT, son identification et son impact. De nouvelles méthodologies améliorées ont été utilisées, en particulier dans l'étude de la phylogénétique , pour valider la présence et l'effet du LGT. Les événements de gain et de duplication de gènes sont équilibrés par la perte de gènes, de sorte que malgré leur nature dynamique, le génome d'une espèce bactérienne reste approximativement de la même taille.
Réarrangement du génome
Les séquences d'insertion génétique mobiles peuvent jouer un rôle dans les activités de réarrangement du génome. Les agents pathogènes qui ne vivent pas dans un environnement isolé contiennent un grand nombre d'éléments de séquence d'insertion et divers segments répétitifs d'ADN. La combinaison de ces deux éléments génétiques est censée aider à la recombinaison homologue . Il existe des agents pathogènes, tels que Burkholderia mallei , et Burkholderia pseudomallei qui présentent des réarrangements à l'échelle du génome en raison de séquences d'insertion et de segments d'ADN répétitifs. À l'heure actuelle, aucune étude ne démontre que des événements de réarrangement à l'échelle du génome donnent directement lieu à un comportement pathogène chez un microbe. Cela ne signifie pas que ce n'est pas possible. Les réarrangements à l'échelle du génome contribuent cependant à la plasticité du génome bactérien, ce qui peut créer les conditions pour que d'autres facteurs introduisent ou perdent des facteurs de virulence.
Polymorphismes mononucléotidiques
Les polymorphismes nucléotidiques simples , ou SNP, permettent une grande variété de variations génétiques chez les humains ainsi que chez les agents pathogènes. Ils permettent aux chercheurs d'estimer une variété de facteurs : les effets des toxines environnementales, la façon dont différentes méthodes de traitement affectent le corps et les causes de la prédisposition d'une personne aux maladies. Les SNP jouent un rôle clé dans la compréhension de la manière dont et pourquoi les mutations se produisent. Les SNP permettent également aux scientifiques de cartographier les génomes et d'analyser les informations génétiques.
Génomes pan et core
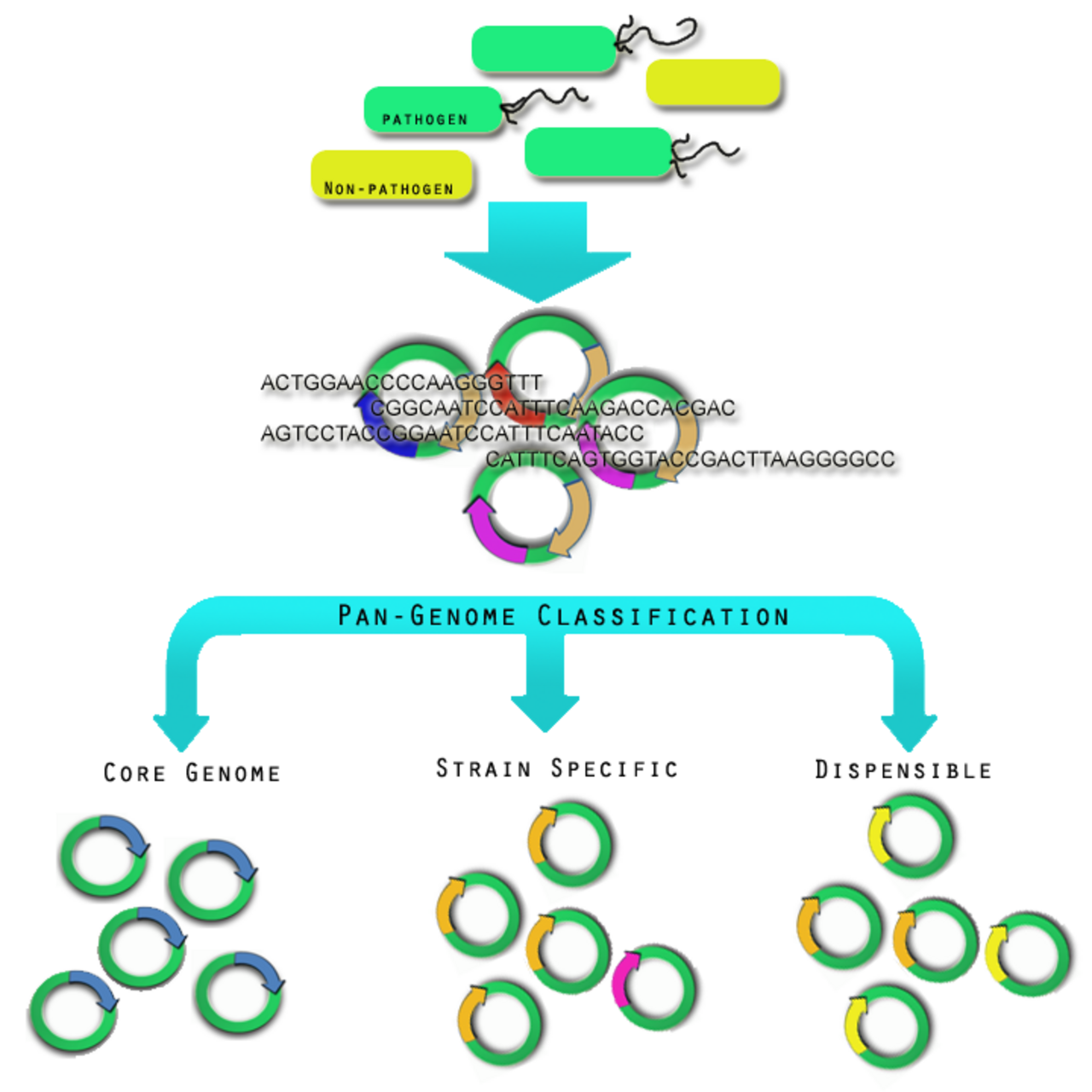
Aperçu du pan-génome La définition la plus récente d'une espèce bactérienne vient de l'ère pré-génomique. En 1987, il a été proposé que les souches bactériennes montrant > 70 % de réassociation ADN·ADN et partageant des traits phénotypiques caractéristiques devraient être considérées comme des souches de la même espèce. La diversité au sein des génomes pathogènes rend difficile l'identification du nombre total de gènes associés à toutes les souches d'une espèce pathogène. On a pensé que le nombre total de gènes associés à une seule espèce pathogène pourrait être illimité, bien que certains groupes tentent d'en déduire une valeur plus empirique. Pour cette raison, il était nécessaire d'introduire le concept de pan-génomes et de génomes centraux. La littérature sur le pan-génome et le génome central a également tendance à avoir un biais en faveur des rapports sur les organismes pathogènes procaryotes. Il faut être prudent lorsqu'on étend la définition d'un pan-génome ou d'un génome central à d'autres organismes pathogènes, car il n'existe aucune preuve formelle des propriétés de ces pan-génomes.
Un génome central est l'ensemble des gènes présents dans toutes les souches d'une espèce pathogène. Un pan-génome est l'ensemble du pool génétique de cette espèce pathogène et comprend des gènes qui ne sont pas partagés par toutes les souches. Les pan-génomes peuvent être ouverts ou fermés selon que l'analyse comparative de plusieurs souches ne révèle aucun nouveau gène (fermé) ou de nombreux nouveaux gènes (ouverts) par rapport au génome central de cette espèce pathogène. Dans le pan-génome ouvert, les gènes peuvent être caractérisés en outre comme dispensables ou spécifiques à la souche. Les gènes dispensables sont ceux trouvés dans plus d'une souche, mais pas dans toutes les souches, d'une espèce pathogène. Les gènes spécifiques à une souche sont ceux trouvés uniquement dans une souche d'une espèce pathogène. Les différences dans les pan-génomes sont le reflet du mode de vie de l'organisme. Par exemple, Streptococcus agalactiae , qui existe dans diverses niches biologiques, possède un pan-génome plus large que Bacillus anthracis, plus isolé sur le plan environnemental . de génomique comparative sont également utilisées pour mieux comprendre le pan-génome. De récentes découvertes montrent que le nombre de nouvelles espèces continue de croître avec environ 10 31 bactériophages sur la planète, ces bactériophages infectant 10 24 autres par seconde, le flux continu de matériel génétique échangé est difficile à imaginer.
Facteurs de virulence
Plusieurs éléments génétiques des agents pathogènes affectant l'homme contribuent au transfert de facteurs de virulence : plasmides , îlots de pathogénicité , prophages , bactériophages, transposons et éléments intégratifs et conjugatifs. Les îlots de pathogénicité et leur détection sont au centre de plusieurs efforts bioinformatiques impliqués dans la pathogénomique. Il est communément admis que les « souches bactériennes environnementales » n'ont pas la capacité de nuire ou de causer des dommages aux humains. Cependant, des études récentes montrent que les bactéries des environnements aquatiques ont acquis des souches pathogènes au cours de l'évolution. Cela permet aux bactéries d'avoir une gamme plus large de traits génétiques et peut constituer une menace potentielle pour les humains, d'où une plus grande résistance aux antibiotiques.
Interactions microbes-microbes
Les interactions microbe-hôte ont tendance à éclipser la prise en compte des interactions microbe-microbe. Les interactions microbe-microbe peuvent cependant conduire à des états d'infirmité chroniques difficiles à comprendre et à traiter.
Biofilms
Les biofilms sont un exemple d'interactions entre microbes et sont considérés comme associés à 80 % des infections humaines. Récemment, il a été démontré que des gènes spécifiques et des protéines de surface cellulaire sont impliqués dans la formation de biofilms. Ces gènes et ces protéines de surface peuvent être caractérisés par des méthodes in silico pour former un profil d'expression des bactéries interagissant avec les biofilms. Ce profil d'expression peut être utilisé dans une analyse ultérieure d'autres microbes pour prédire le comportement des microbes des biofilms ou pour comprendre comment démanteler la formation des biofilms.
Analyse du microbe hôte
Les agents pathogènes ont la capacité de s'adapter et de manipuler les cellules hôtes, en tirant pleinement parti des processus et mécanismes cellulaires d'une cellule hôte.
Un microbe peut être influencé par ses hôtes pour s'adapter à son nouvel environnement ou apprendre à l'éviter. Une compréhension de ces comportements fournira des informations utiles pour des thérapies potentielles. Le programme européen de recherche sur la pathogénomique présente les initiatives les plus détaillées sur les interactions entre l'hôte et le microbe. Son rapport met l'accent sur les caractéristiques suivantes :
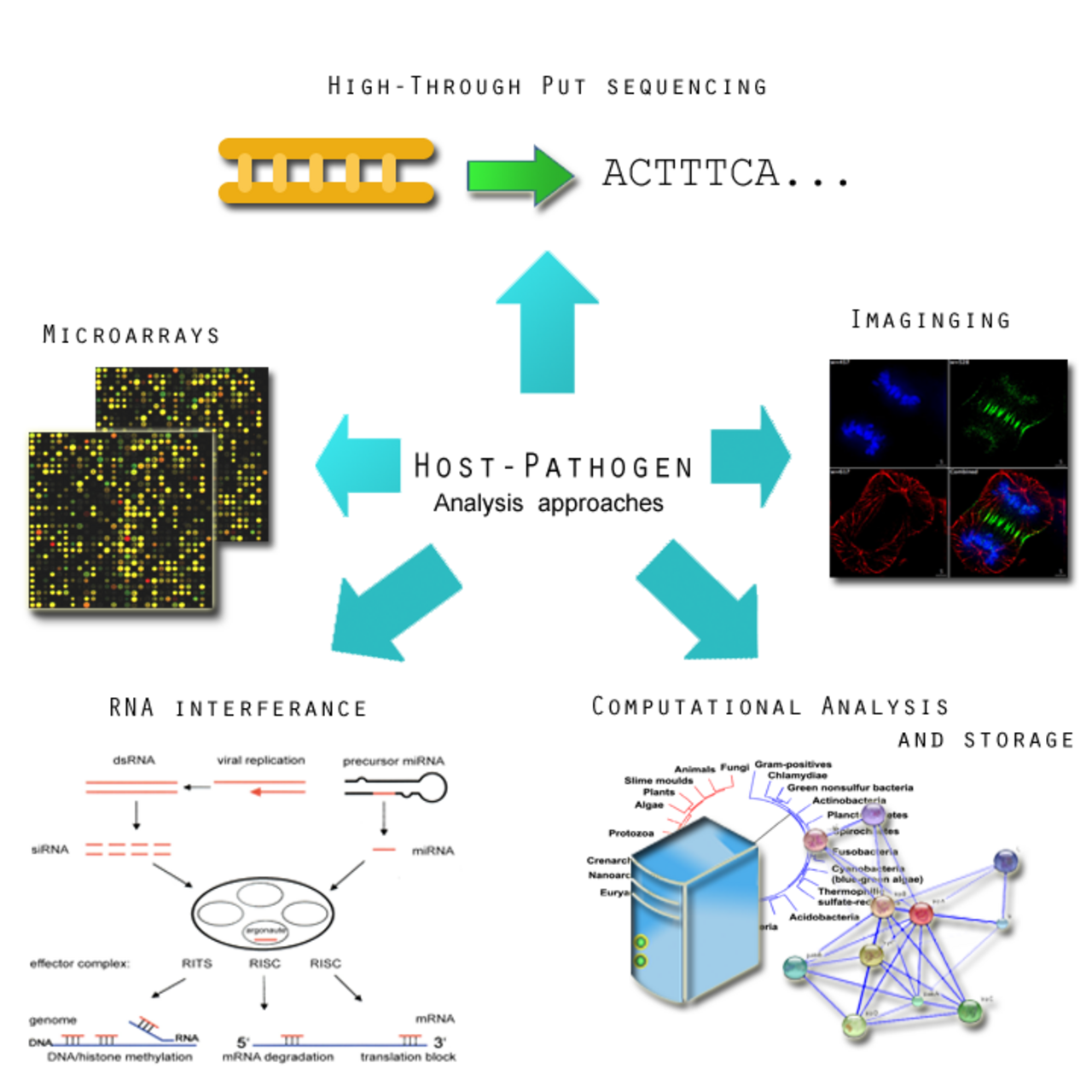
- Analyse par microarray de l'expression des gènes de l'hôte et du microbe pendant l'infection . Ceci est important pour identifier l'expression des facteurs de virulence qui permettent à un pathogène de survivre au mécanisme de défense d'un hôte. Les pathogènes ont tendance à subir un assortiment de changements afin de subvertir le système immunitaire de l'hôte, favorisant dans certains cas un état génomique hyper variable. Les études d'expression génomique seront complétées par des études de réseaux d'interaction protéine-protéine.
- Utilisation de l'interférence ARN (ARNi) pour identifier les fonctions des cellules hôtes en réponse aux infections . L'infection dépend de l'équilibre entre les caractéristiques de la cellule hôte et de la cellule pathogène. Dans certains cas, il peut y avoir une réponse hyperactive de l'hôte à l'infection, comme dans la méningite, qui peut submerger l'organisme de l'hôte. En utilisant l'ARN, il sera possible d'identifier plus clairement comment une cellule hôte se défend en cas d'infection aiguë ou chronique. Cette méthode a également été appliquée avec succès chez la drosophile.
- Les interactions entre les microbes dans l'environnement hôte ne sont pas toutes malveillantes. La flore commensale , présente dans divers environnements chez les animaux et les humains, peut en fait aider à combattre les infections microbiennes. La flore humaine , comme l'intestin par exemple, abrite une myriade de microbes.
La diversité de la communauté intestinale est considérée comme essentielle à la santé humaine. Plusieurs projets sont en cours pour mieux comprendre les écosystèmes intestinaux. La séquence de la souche commensale d'Escherichia coli SE11, par exemple, a déjà été déterminée à partir des matières fécales d'un humain en bonne santé et promet d'être la première d'une longue série d'études. Grâce à l'analyse génomique et à l'analyse ultérieure des protéines, une meilleure compréhension des propriétés bénéfiques de la flore commensale sera étudiée dans l'espoir de comprendre comment développer un meilleur traitement.
Perspective éco-évolutive
La perspective « éco-évolution » des interactions pathogène-hôte met l'accent sur les influences de l'écologie et de l'environnement sur l'évolution du pathogène. Les facteurs génomiques dynamiques tels que la perte de gènes, le gain de gènes et le réarrangement du génome sont tous fortement influencés par les changements dans la niche écologique où réside une souche microbienne particulière. Les microbes peuvent passer d'un état pathogène à un état non pathogène en raison de changements d'environnement. Cela a été démontré lors d'études sur la peste, Yersinia pestis , qui a apparemment évolué d'un pathogène gastro-intestinal léger à un microbe très hautement pathogène par le biais d'événements génomiques dynamiques. Pour que la colonisation se produise, il doit y avoir des changements dans la composition biochimique pour aider à la survie dans divers environnements. Cela est très probablement dû à un mécanisme permettant à la cellule de détecter les changements dans l'environnement, influençant ainsi le changement dans l'expression des gènes. Comprendre comment ces changements de souche se produisent, passant d'un état faiblement ou non pathogène à un état hautement pathogène et vice versa, peut aider à développer de nouvelles thérapies pour les infections microbiennes.
Applications
La santé humaine s’est grandement améliorée et le taux de mortalité a considérablement diminué depuis la Seconde Guerre mondiale en raison d’une meilleure hygiène due à l’évolution des réglementations de santé publique, ainsi que de vaccins et d’antibiotiques plus facilement disponibles. La pathogénomique permettra aux scientifiques d’élargir leurs connaissances sur les microbes pathogènes et non pathogènes, ce qui permettra de développer de nouveaux vaccins améliorés. La pathogénomique a également des implications plus larges, notamment en matière de prévention du bioterrorisme.
Vaccinologie inverse
La vaccinologie inverse est relativement nouvelle. Bien que la recherche soit encore en cours, des percées ont été réalisées avec des agents pathogènes tels que le streptocoque et la méningite . Les méthodes de production de vaccins, telles que les méthodes biochimiques et sérologiques, sont laborieuses et peu fiables. Elles nécessitent que les agents pathogènes soient in vitro pour être efficaces. Les nouvelles avancées dans le développement génomique aident à prédire presque toutes les variations des agents pathogènes, ce qui fait progresser les vaccins. Des vaccins à base de protéines sont en cours de développement pour lutter contre les agents pathogènes résistants tels que le staphylocoque et la chlamydia .
Lutte contre le bioterrorisme
En 2005, la séquence de la grippe espagnole de 1918 a été achevée. Une analyse phylogénétique a permis de fournir un compte rendu détaillé de l'évolution et du comportement du virus, en particulier de son adaptation à l'homme. Après le séquençage de la grippe espagnole, le pathogène a également été reconstitué. Une fois inséré dans des souris, le pathogène s'est révélé incroyablement mortel. Les attaques à l'anthrax de 2001 ont mis en lumière la possibilité que le bioterrorisme soit une menace plus réelle qu'imaginée. Le bioterrorisme avait été anticipé lors de la guerre en Irak, avec des soldats vaccinés contre la variole . En utilisant les technologies et les connaissances acquises grâce à la reconstitution de la grippe espagnole, il pourrait être possible d'empêcher de futures épidémies mortelles de maladies implantées. Il existe cependant une forte préoccupation éthique quant à savoir si la résurrection de vieux virus est nécessaire et si elle fait plus de mal que de bien. La meilleure façon de contrer de telles menaces est de se coordonner avec les organisations qui fournissent des vaccins. Une sensibilisation et une participation accrues diminueraient considérablement l'efficacité d'une éventuelle épidémie. Une mesure supplémentaire consisterait à surveiller les réservoirs d'eau naturels afin de prévenir une attaque ou une épidémie. Dans l'ensemble, la communication entre les laboratoires et les grandes organisations, telles que le Global Outbreak Alert and Response Network (GOARN), peut conduire à une détection précoce et à la prévention des épidémies.