L'une des principales fonctions des plaquettes est de contribuer à l'hémostase : le processus d'arrêt du saignement au niveau de l'interruption de la paroi interne des vaisseaux ( endothélium ). Les plaquettes s'agglutinent à cet endroit et, sauf si l'interruption est trop importante, elles la bouchent. Premièrement, les plaquettes se fixent à des substances situées à l'extérieur de l'endothélium interrompu : adhésion . Deuxièmement, elles changent de forme, activent des récepteurs et sécrètent des messagers chimiques : activation . Troisièmement, elles s'agrègent entre elles par l'intermédiaire de ponts récepteurs : agrégation . La formation de ce clou plaquettaire (hémostase primaire) est associée à l'activation de la cascade de coagulation , avec pour conséquence le dépôt et la formation de fibrine (hémostase secondaire). Ces processus peuvent se chevaucher : on peut observer un clou principalement plaquettaire, ou « caillot blanc », un clou principalement fibrineux, ou « caillot rouge », ou encore un mélange plus fréquent. Berridge ajoute la rétraction et l'inhibition plaquettaire comme quatrième et cinquième étapes, tandis que d'autres y ajoutent une sixième étape : la cicatrisation . Les plaquettes participent aux réponses immunitaires intravasculaires innées et adaptatives .
En plus de faciliter le processus de coagulation, les plaquettes contiennent des cytokines et des facteurs de croissance qui peuvent favoriser la cicatrisation des plaies et la régénération des tissus endommagés.
glycoprotéines nécessaires à l'adhésion, à l'activation et à l'agrégation plaquettaires. Par exemple : GPIb/IX/V ; GPVI ; GPIIb/IIIaForme
Les plaquettes inactivées circulantes sont des structures discoïdes biconvexes (en forme de lentille), 2–3 μm de diamètre maximal. Les plaquettes activées ont des projections de membrane cellulaire recouvrant leur surface.
En première approximation, la forme peut être assimilée à celle d' un sphéroïde aplati , avec un rapport des demi-axes de 2 à 8 Cette approximation permet de modéliser les propriétés hydrodynamiques et optiques d'une population, ainsi que de reconstituer les paramètres géométriques de plaquettes individuelles mesurées par cytométrie en flux . Des modèles biophysiques plus précis de la morphologie de surface des plaquettes, modélisant leur forme à partir des principes fondamentaux, permettent d'obtenir une géométrie plaquettaire plus réaliste à l'état basal et activé
Développement
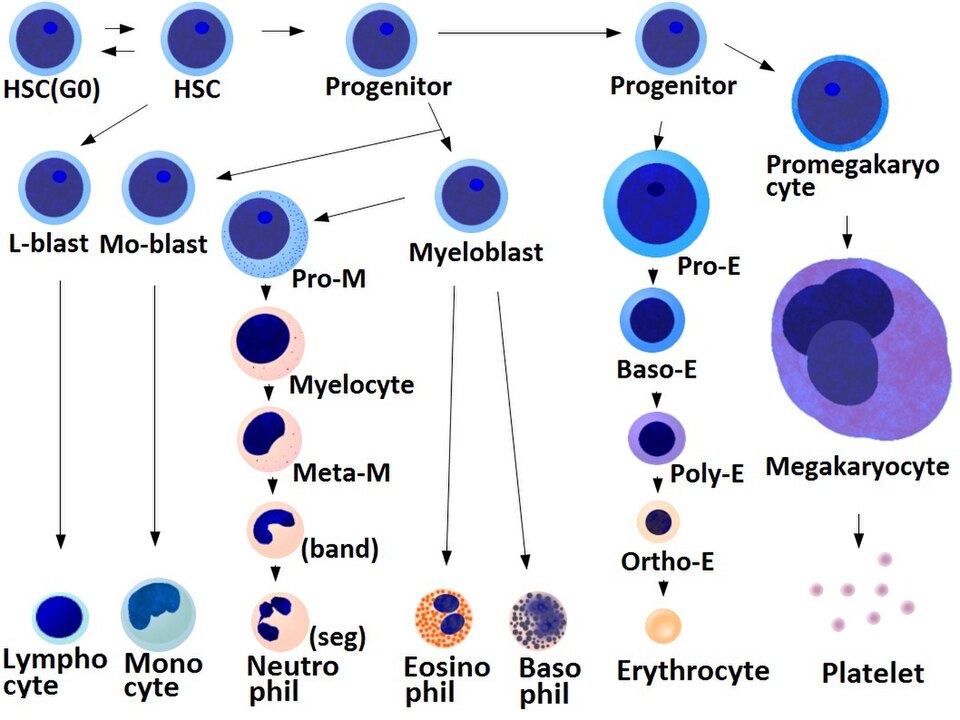
- La production de mégacaryocytes et de plaquettes est régulée par la thrombopoïétine , une hormone produite dans les reins et le foie.
- Chaque mégacaryocyte produit entre 1 000 et 3 000 plaquettes au cours de sa vie.
- Un adulte en bonne santé produit en moyenne 10<sup> 11 </sup> plaquettes par jour.
- Les plaquettes de réserve sont stockées dans la rate et sont libérées en cas de besoin par la contraction splénique induite par le système nerveux sympathique.
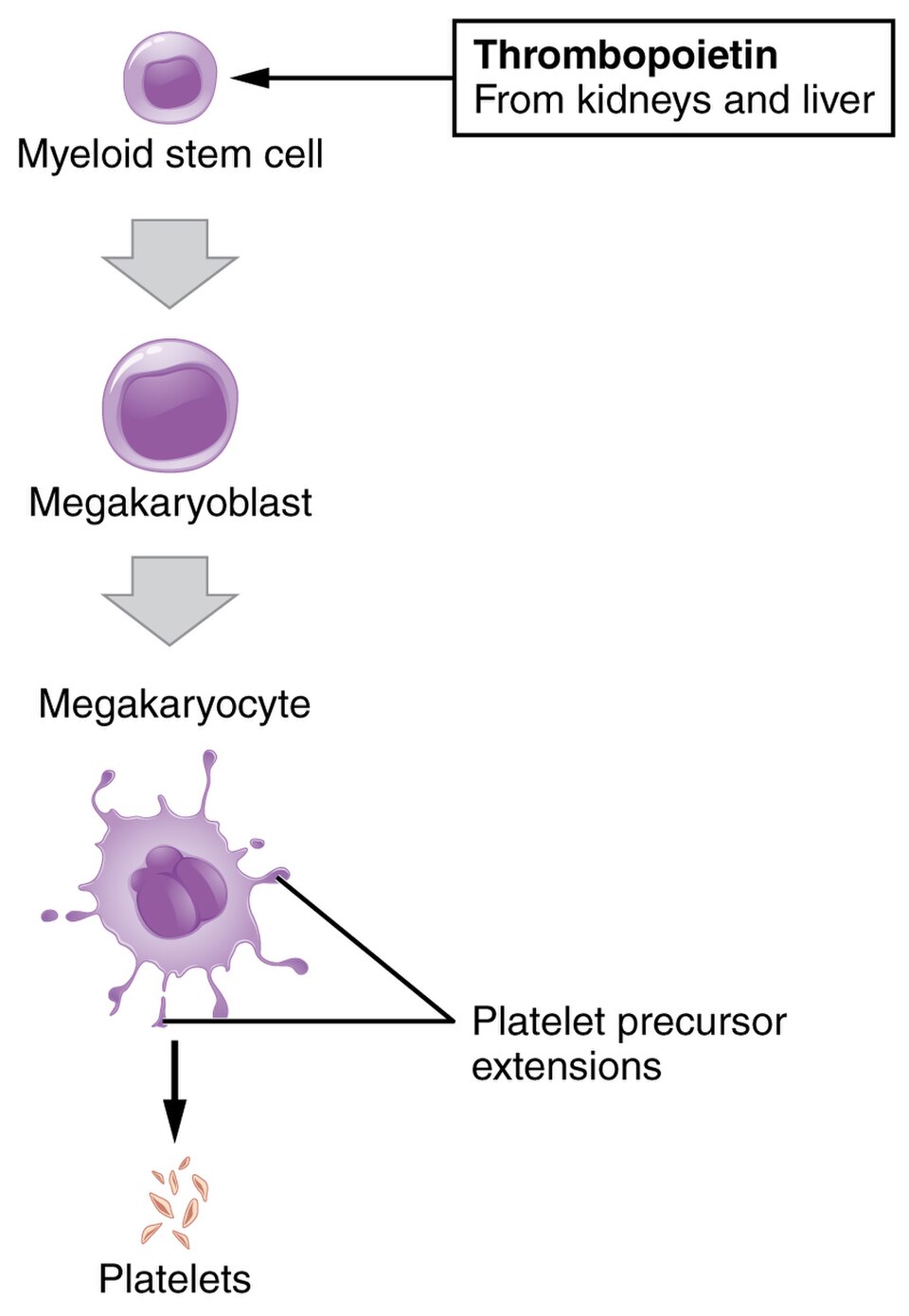
- La durée de vie moyenne des plaquettes circulantes est de 8 à 9 jours. La durée de vie des plaquettes individuelles est contrôlée par la voie de régulation apoptotique interne, qui possède une minuterie Bcl-x L.
- Les vieilles plaquettes sont détruites par phagocytose dans la rate et le foie.
Hémostase
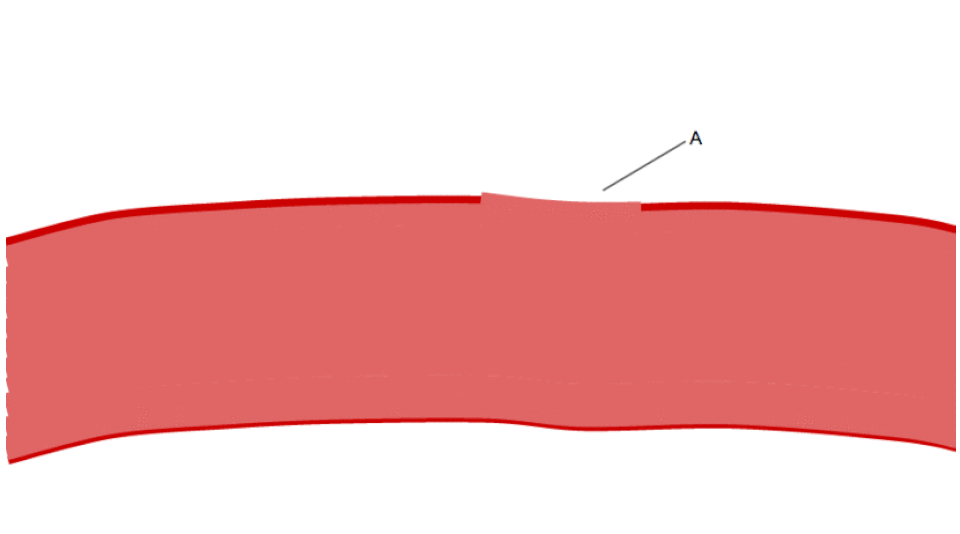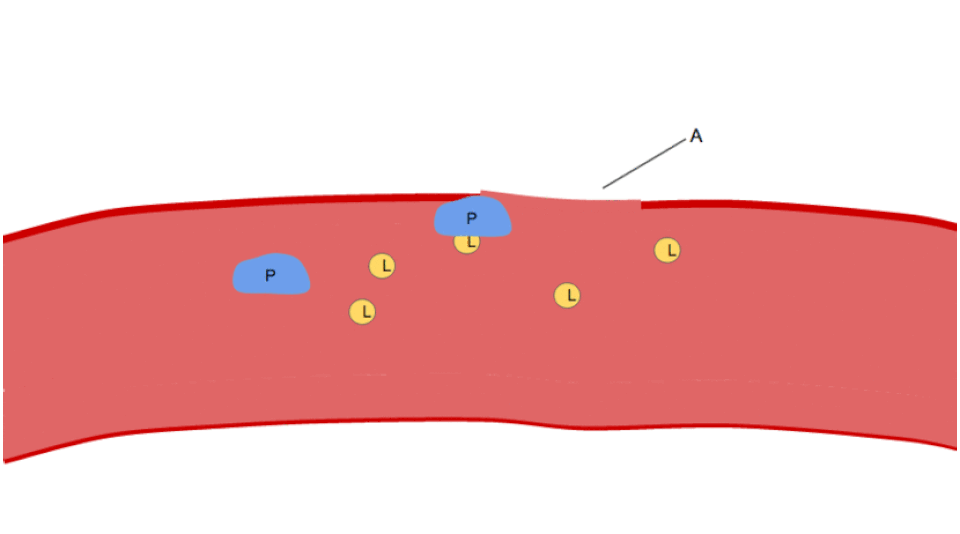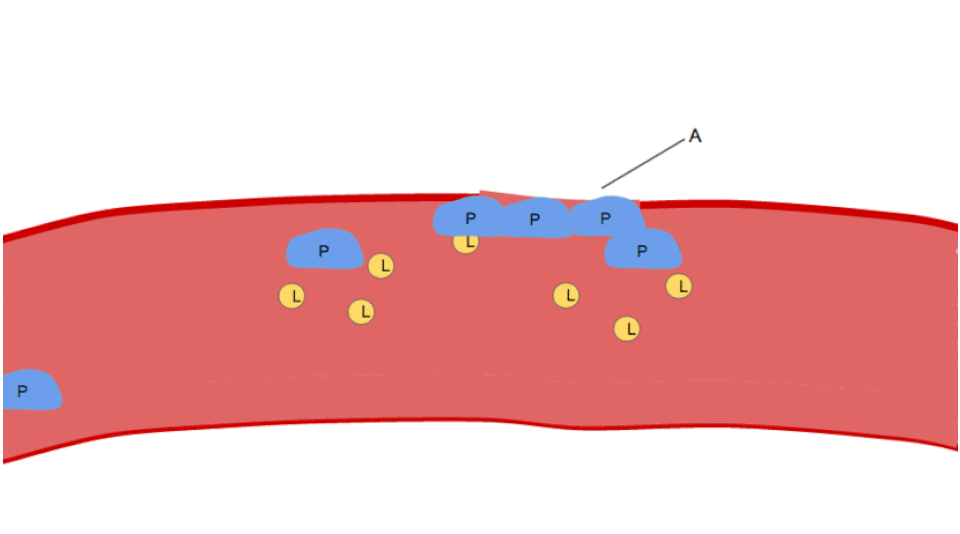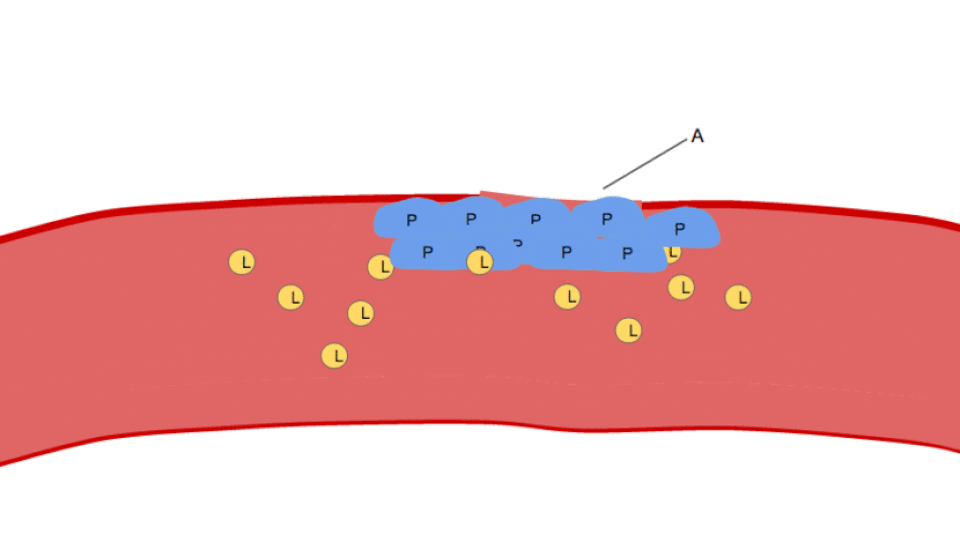
La fonction fondamentale des plaquettes est de s'agréger pour arrêter un saignement aigu. Ce processus est complexe, car plus de 193 protéines et 301 interactions sont impliquées dans la dynamique plaquettaire. Malgré de nombreux chevauchements, la fonction plaquettaire peut être modélisée en trois étapes :
Adhésion
La formation de thrombus sur un endothélium intact est empêchée par l'oxyde nitrique , la prostacycline , et le CD39 .
Les cellules endothéliales adhèrent au collagène sous-endothélial grâce au facteur de von Willebrand (VWF), qu'elles produisent. Le VWF est également stocké dans les corps de Weibel-Palade des cellules endothéliales et sécrété de façon constitutive dans le sang. Les plaquettes stockent le VWF dans leurs granules alpha.
Lorsque la couche endothéliale est perturbée, le collagène et le VWF ancrent les plaquettes au sous-endothélium. Le récepteur plaquettaire GP1b-IX-V se lie au VWF ; et le récepteur GPVI et l’intégrine α2β1 se lient au collagène.
Activation
Inhibition
Des facteurs provenant de la paroi des vaisseaux empêchent l'activation des plaquettes. Une paroi endothéliale intacte inhibe l'activation plaquettaire en produisant de l'oxyde nitrique , de l'ADPase endothéliale et de la PGI₂ (prostacycline). L'ADPase endothéliale dégrade l' ADP, activateur plaquettaire .
Les plaquettes au repos maintiennent un efflux actif de calcium via une pompe à calcium activée par l'AMPc . La concentration intracellulaire de calcium détermine l'état d'activation plaquettaire, car elle agit comme second messager , induisant un changement de conformation et une dégranulation plaquettaires. La prostacycline endothéliale se lie aux récepteurs des prostanoïdes à la surface des plaquettes au repos. Cette liaison stimule la protéine Gs couplée , ce qui augmente l'activité de l'adénylate cyclase et la production d'AMPc, favorisant ainsi l'efflux de calcium et réduisant la disponibilité du calcium intracellulaire nécessaire à l'activation plaquettaire.
L'ADP se lie aux récepteurs purinergiques à la surface des plaquettes. Le récepteur purinergique thrombocytaire P2Y12 étant couplé aux protéines Gi , l'ADP réduit l'activité de l'adénylate cyclase plaquettaire et la production d'AMPc, ce qui entraîne une accumulation de calcium intracellulaire par inactivation de la pompe d'efflux de calcium-AMPc. L'autre récepteur de l'ADP, P2Y1 , est couplé à la protéine Gq, qui active la phospholipase C-β2 ( PLCB2 ), induisant la production d'inositol 1,4,5-trisphosphate (IP3) et la libération intracellulaire de calcium. L'ensemble de ces mécanismes induit l'activation plaquettaire. L'ADPase endothéliale dégrade l'ADP et empêche ce processus. Le clopidogrel et les antiagrégants plaquettaires apparentés agissent également comme antagonistes du récepteur purinergique P2Y12 . Les données suggèrent que l'ADP active la voie PI3K/Akt lors d'une première vague d'agrégation, conduisant à la génération de thrombine et à l'activation de PAR-1 , ce qui provoque une deuxième vague d'agrégation.
Déclencheur (induction)
L’activation plaquettaire débute quelques secondes après l’adhésion. Elle est déclenchée lorsque le collagène du sous-endothélium se lie à ses récepteurs ( récepteur GPVI et intégrine α2β1) sur la plaquette. Le GPVI est associé à la chaîne gamma du récepteur Fc et conduit, via l’activation d’une cascade de tyrosine kinase, à l’activation de la PLC-gamma2 ( PLCG2 ) et à une libération accrue de calcium.
Le facteur tissulaire se lie également au facteur VII dans le sang, ce qui initie la cascade de coagulation extrinsèque et augmente la production de thrombine . La thrombine est un puissant activateur plaquettaire, agissant via les récepteurs couplés aux protéines G Gq et G12. Ces récepteurs activent des voies de signalisation dépendantes du calcium au sein de la plaquette, inhibant ainsi l'efflux basal de calcium. Les trois familles de protéines G (Gq, Gi et G12) agissent de concert pour une activation complète. La thrombine favorise également le renforcement secondaire du clou plaquettaire par la fibrine . L'activation plaquettaire induit la dégranulation et la libération du facteur V et du fibrinogène , potentialisant ainsi la cascade de coagulation. L'agrégation plaquettaire et la coagulation se produisent simultanément, chacune induisant l'autre pour former le thrombus final réticulé par la fibrine.
Composantes (conséquences)
Activation de GPIIb/IIIa
La signalisation via le GPVI, médiée par le collagène, augmente la production plaquettaire de thromboxane A2 (TXA2) et diminue celle de prostacycline . Ce phénomène résulte d'une modification du flux métabolique de la voie de synthèse des eicosanoïdes plaquettaires , impliquant les enzymes phospholipase A2 , cyclo-oxygénase 1 et thromboxane A synthase . Les plaquettes sécrètent du thromboxane A2, qui agit sur leurs propres récepteurs à la surface plaquettaire (d'où le mécanisme dit « de l'extérieur vers l'intérieur ») ainsi que sur ceux d'autres plaquettes. Ces récepteurs déclenchent une signalisation intraplaquettaire, qui active les récepteurs GPIIb/IIIa pour initier l'agrégation plaquettaire .
Sécrétion granulaire
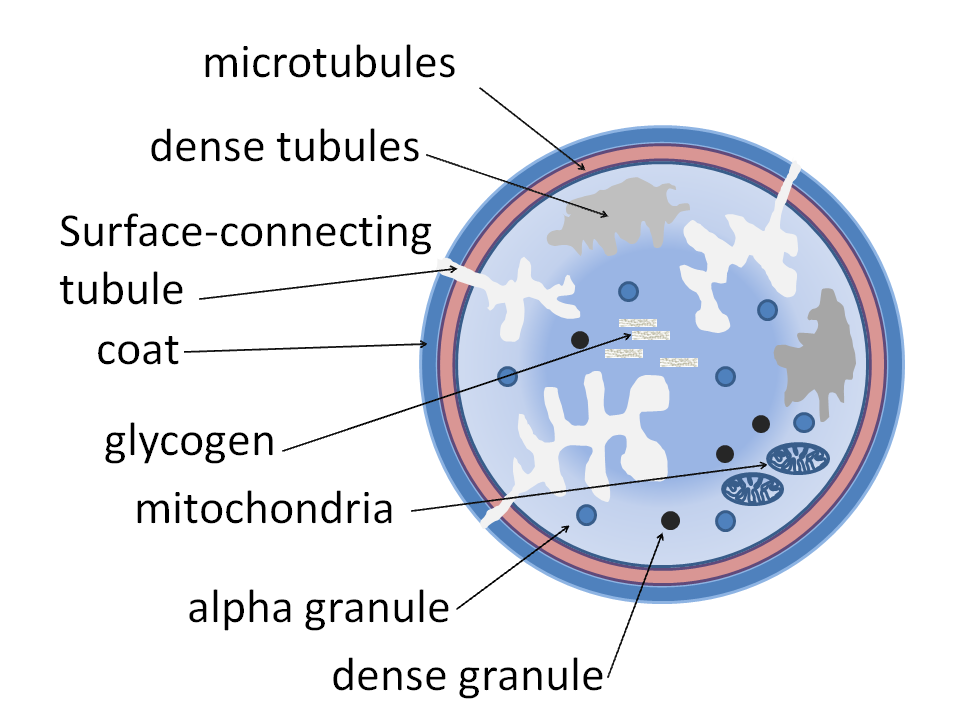
Les plaquettes contiennent des granules denses , des granules lambda et des granules alpha . Les plaquettes activées sécrètent le contenu de ces granules par leurs canaux semi-conducteurs vers l'extérieur. Les plaquettes liées et activées dégranulent pour libérer des agents chimiotactiques plaquettaires , attirant ainsi davantage de plaquettes sur le site de la lésion endothéliale. Caractéristiques des granules :
- Granules α (granules alpha) — contenant la P-sélectine , le facteur plaquettaire 4 , le facteur de croissance transformant β1 , le facteur de croissance dérivé des plaquettes , la fibronectine , la β-thromboglobuline , le facteur von Willebrand (vWF) , le fibrinogène et les facteurs de coagulation V et XIII.
- Granules δ (granules delta ou denses) — contenant de l'ADP ou de l'ATP , du calcium et de la sérotonine
- Granules γ (granules gamma) — semblables aux lysosomes et contenant plusieurs enzymes hydrolytiques
- Granules λ (granules lambda) — contenus impliqués dans la résorption lors des stades ultérieurs de la réparation vasculaire
Changement morphologique
Comme l'ont montré la cytométrie en flux et la microscopie électronique , le signe le plus sensible d'activation plaquettaire, après exposition des plaquettes à l'ADP, est une modification morphologique . L'hyperpolarisation mitochondriale est un événement clé dans l'initiation de ces modifications . La concentration de calcium intraplaquettaire augmente, stimulant l'interaction entre le complexe microtubule/filament d'actine. Les changements de forme continus, de la plaquette non activée à la plaquette pleinement activée, sont particulièrement visibles en microscopie électronique à balayage . Les trois étapes de ce processus sont appelées phase dendritique précoce , phase d'étalement précoce et phase d'étalement . La surface de la plaquette non activée ressemble à celle du cerveau : un aspect ridé dû à de nombreux replis superficiels qui augmentent sa surface ; la phase dendritique précoce , à une pieuvre aux multiples bras et jambes ; la phase d'étalement précoce , à un œuf cru au plat, dont le « jaune » constitue le corps central ; et la phase d'étalement , à un œuf cuit au plat avec un corps central plus dense.
Ces modifications résultent toutes de l'interaction du complexe microtubule/actine avec la membrane plaquettaire et le système canaliculaire ouvert (SCO), qui correspond à une extension et une invagination de cette membrane. Ce complexe, situé juste sous ces membranes, agit comme un moteur chimique qui expulse le SCO invaginé de l'intérieur de la plaquette, à la manière de retourner les poches d'un pantalon, créant ainsi les dendrites. Ce processus est similaire au mécanisme de contraction d'une cellule musculaire . Le SCO devient alors totalement indiscernable de la membrane plaquettaire initiale, formant ainsi la structure en « œuf au plat ». Cette augmentation spectaculaire de la surface se produit sans étirement ni ajout de phospholipides à la membrane plaquettaire.
Interactions plaquettes-facteurs de coagulation : facilitation de la coagulation
L'activation plaquettaire entraîne une charge négative à la surface de la membrane plaquettaire. Une des voies de signalisation active la scramblase , qui transfère les phospholipides chargés négativement de la face interne à la face externe de la membrane plaquettaire. Ces phospholipides se lient ensuite aux complexes ténase et prothrombinase , deux sites d'interaction entre les plaquettes et la cascade de coagulation. Les ions calcium sont indispensables à la liaison de ces facteurs de coagulation.
Outre leur interaction avec le facteur von Willebrand (vWF) et la fibrine, les plaquettes interagissent avec la thrombine, les facteurs X, Va, VIIa, XI, IX et la prothrombine pour achever leur formation via la cascade de coagulation. Les plaquettes humaines n'expriment pas le facteur tissulaire . Les plaquettes de rat expriment la protéine du facteur tissulaire et portent à la fois l'ARN pré-messager et l'ARN messager mature du facteur tissulaire.
Agrégation
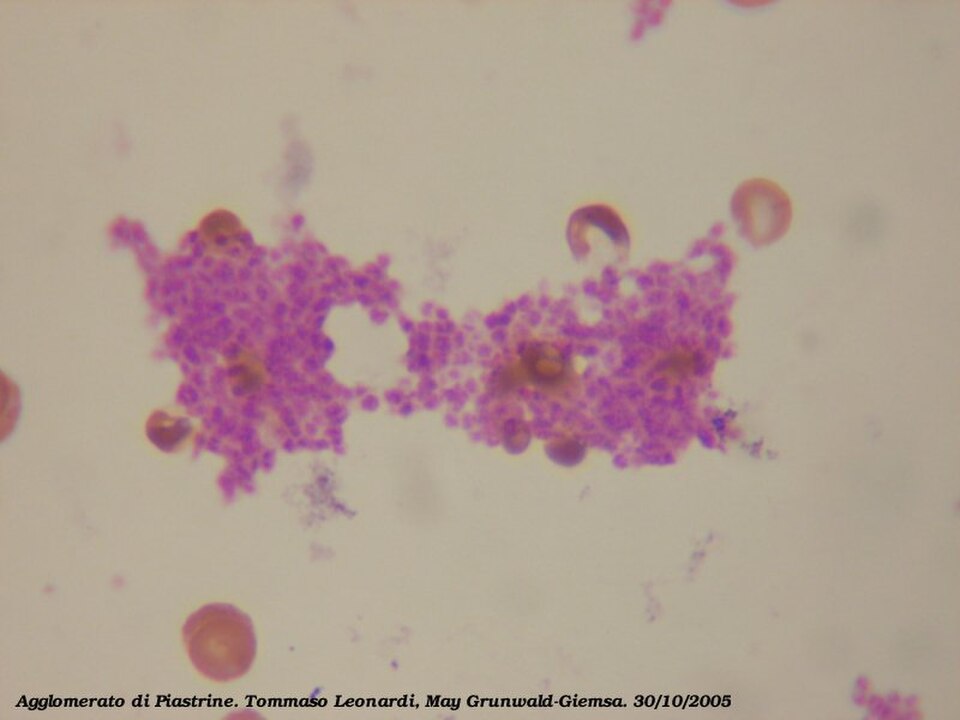
L'agrégation plaquettaire débute quelques minutes après l'activation et résulte de l'activation du récepteur GPIIb/IIIa , permettant à ce dernier de se lier au facteur von Willebrand (vWF) ou au fibrinogène . Chaque plaquette possède environ 60 000 de ces récepteurs. Lors de l'activation plaquettaire, lorsqu'un ou plusieurs des neuf récepteurs de surface plaquettaires différents sont activés, les voies de signalisation intraplaquettaires induisent un changement de conformation des récepteurs GPIIb/IIIa existants (de courbés à rectilignes), les rendant ainsi capables de se lier.
Le fibrinogène étant une protéine en forme de bâtonnet présentant des nodules à chaque extrémité capables de se lier au GPIIb/IIIa, les plaquettes activées dont le GPIIb/IIIa est exposé peuvent fixer le fibrinogène pour former des agrégats. Le GPIIb/IIIa peut également ancrer les plaquettes au facteur von Willebrand sous-endothélial, assurant ainsi une stabilisation structurale supplémentaire.
Classiquement, on pensait que c'était le seul mécanisme impliqué dans l'agrégation, mais trois autres mécanismes ont été identifiés qui peuvent initier l'agrégation, en fonction de la vitesse du flux sanguin (c'est-à-dire de la plage de cisaillement).
Fonction immunitaire
Les plaquettes jouent un rôle central dans l'immunité innée , en initiant et en participant à de nombreux processus inflammatoires, en se liant directement aux agents pathogènes et même en les détruisant. Les données cliniques montrent que de nombreux patients atteints d'infections bactériennes ou virales graves présentent une thrombocytopénie , ce qui réduit leur contribution à l'inflammation. Les agrégats plaquettes-leucocytes (APL) présents dans la circulation sont typiques de la septicémie ou des maladies inflammatoires de l'intestin , illustrant le lien entre les thrombocytes et les cellules immunitaires.
La membrane des plaquettes possède des récepteurs au collagène. Suite à la rupture de la paroi du vaisseau sanguin, les plaquettes sont exposées et adhèrent au collagène des tissus environnants.
Immunothrombose
L'hémostase étant une fonction essentielle des thrombocytes chez les mammifères, elle joue également un rôle dans le confinement des infections. En cas de blessure, les plaquettes, associées à la cascade de coagulation, constituent la première ligne de défense en formant un caillot sanguin. L'hémostase et la défense de l'hôte sont donc étroitement liées au cours de l'évolution. Par exemple, chez la limule de l'Atlantique (dont l'âge est estimé à plus de 400 millions d'années), l'unique type de cellule sanguine, l' amœbocyte , assure à la fois l'hémostase et les fonctions immunitaires, notamment l'encapsulation et la phagocytose des pathogènes , ainsi que l'exocytose des granules intracellulaires contenant des molécules de défense bactéricides . La coagulation sanguine soutient la fonction immunitaire en piégeant les bactéries.
La thrombose (coagulation sanguine dans les vaisseaux intacts) est généralement considérée comme une réponse immunitaire pathologique, entraînant l'obstruction de la lumière vasculaire et des lésions tissulaires hypoxiques. Dans certains cas, cependant, la thrombose dirigée (ou immunothrombose) peut contrôler localement la propagation d'une infection. Cette thrombose est dirigée en coordination avec les plaquettes, les neutrophiles et les monocytes . Le processus est initié soit par les cellules immunitaires via l'activation de leurs récepteurs de reconnaissance de motifs (PRR), soit par la liaison plaquettes-bactéries. Les plaquettes peuvent se lier aux bactéries soit directement par l'intermédiaire des PRR thrombocytaires et des protéines de surface bactériennes, soit via des protéines plasmatiques qui se lient à la fois aux plaquettes et aux bactéries . Les monocytes répondent aux motifs moléculaires associés aux pathogènes (PAMP) ou aux motifs moléculaires associés aux dommages (DAMP) bactériens en activant la voie extrinsèque de la coagulation. Les neutrophiles favorisent la coagulation sanguine par NETose , tandis que les plaquettes favorisent la NETose des neutrophiles. Les NETs se lient au facteur tissulaire, fixant ainsi les centres de coagulation au site de l'infection. Ils activent également la voie intrinsèque de la coagulation en fournissant une surface chargée négativement au facteur XII. D'autres sécrétions de neutrophiles, telles que les enzymes protéolytiques qui clivent les inhibiteurs de la coagulation, renforcent également ce processus.
En cas de déséquilibre dans la régulation de l'immunothrombose, ce processus peut devenir aberrant. On soupçonne que des anomalies de régulation de l'immunothrombose constituent un facteur majeur de thrombose pathologique, notamment dans des formes telles que la coagulation intravasculaire disséminée (CIVD) ou la thrombose veineuse profonde . La CIVD associée à la septicémie illustre parfaitement à la fois le dérèglement du processus de coagulation et une réponse inflammatoire systémique excessive. Elle entraîne la formation d'une multitude de microthrombi, dont la composition est similaire à celle des thrombi produits lors d'une immunothrombose native : ils sont composés de fibrine, de plaquettes, de neutrophiles et de NET (pièges extracellulaires de neutrophiles).
Inflammation
Les plaquettes migrent rapidement vers les sites de lésion ou d'infection. Elles y moduleraient les processus inflammatoires par le biais d'interactions avec les leucocytes et de la sécrétion de cytokines , de chimiokines et d'autres médiateurs inflammatoires. Les plaquettes sécrètent également le facteur de croissance dérivé des plaquettes (PDGF).
Les plaquettes modulent les neutrophiles en formant des agrégats plaquettes-leucocytes (APL). Ces agrégats induisent une augmentation de la production du récepteur du complément αmβ2 ( Mac-1 ) par les neutrophiles. L'interaction avec les APL induit également la dégranulation et une phagocytose accrue des neutrophiles.
Les plaquettes constituent la principale source de CD40L soluble (CD154), qui induit la production d' espèces réactives de l'oxygène (ROS) et augmente l'expression des molécules d'adhésion (telles que la sélectine E , l'ICAM-1 et la VCAM-1 ) dans les neutrophiles. Le CD40L active également les macrophages et la réponse cytotoxique des T et B. [
Les plaquettes de mammifères, dépourvues de noyau, sont capables de se déplacer de manière autonome. Elles agissent comme des cellules phagocytaires actives, érodant la paroi des vaisseaux sanguins et réorganisant le thrombus. Elles peuvent reconnaître et adhérer à de nombreuses surfaces, y compris les bactéries, et les envelopper dans leur système canaliculaire ouvert (SCO). Ce processus a été proposé sous le nom de covercytose (OCS) plutôt que de phagocytose, car l'OCS correspond à une simple invagination de la membrane plasmique externe. Ces complexes plaquettes-bactéries constituent une plateforme d'interaction pour les neutrophiles, qui détruisent les bactéries par NET et phagocytose.
Les plaquettes participent également aux maladies inflammatoires chroniques, telles que la synovite ou la polyarthrite rhumatoïde . Elles sont activées par la glycoprotéine IV (GPVI), un récepteur du collagène. Les microvésicules plaquettaires pro-inflammatoires induisent une sécrétion continue de cytokines par les synoviocytes voisins , de type fibroblastique , notamment l'IL-6 et l'IL-8 . Les lésions inflammatoires de la matrice extracellulaire environnante exposent continuellement davantage de collagène, qui se lie aux récepteurs plaquettaires et maintient la production de microvésicules.
Immunité adaptative
Les plaquettes activées participent à l'immunité adaptative en interagissant avec les anticorps . Elles se lient spécifiquement aux IgG via le FcγRIIA , un récepteur du fragment constant (Fc) des IgG. Une fois activées et liées à des bactéries opsonisées par les IgG , les plaquettes libèrent des espèces réactives de l'oxygène (ROS), des peptides antimicrobiens, des défensines , des kinocidines et des protéases , induisant ainsi la destruction directe des bactéries. Les plaquettes sécrètent également des médiateurs pro-inflammatoires et procoagulants, tels que les polyphosphates inorganiques ou le facteur plaquettaire 4 (PF4), assurant la liaison entre les réponses immunitaires innées et adaptatives.
Mesure et essais
Mesures
La concentration plaquettaire dans le sang (c.-à-d. le nombre de plaquettes) peut être mesurée manuellement à l'aide d'un hémocytomètre ou en plaçant le sang dans un analyseur de plaquettes automatisé utilisant le comptage de particules, tel qu'un compteur Coulter ou des méthodes optiques. La plupart des méthodes d'analyse sanguine courantes incluent le nombre de plaquettes dans leurs mesures, généralement exprimé en PLT .
La concentration plaquettaire varie d'un individu à l'autre et au fil du temps, la moyenne dans la population se situant entre 250 000 et 260 000 cellules par mm³ ( soit par microlitre), mais la plage normale généralement acceptée en laboratoire se situe entre 150 000 et 400 000 cellules par mm³ ou 150 à 400 milliards par litre.

Sur un frottis sanguin coloré , les plaquettes apparaissent comme des taches violet foncé, d'environ 20 % du diamètre des globules rouges. Le frottis permet d'observer leur taille, leur forme, leur nombre et leur agrégation . Un adulte en bonne santé possède généralement 10 à 20 fois plus de globules rouges que de plaquettes.
Temps de saignement
Le temps de saignement a été mis au point par Duke en 1910 comme test de la fonction plaquettaire. Ce test mesurait le temps nécessaire à l'arrêt du saignement d'une plaie standardisée au lobe de l'oreille, tamponnée toutes les 30 secondes ; un temps inférieur à 3 minutes était considéré comme normal. Le temps de saignement présente une faible sensibilité et spécificité pour les troubles plaquettaires légers à modérés et n'est plus recommandé pour le dépistage.
Agrégométrie à électrodes multiples
le plasma riche en plaquettes est placé entre une source lumineuse et une cellule photoélectrique . Le plasma non agrégé laisse passer relativement peu de lumière. Après l'ajout d'un agoniste, les plaquettes s'agrègent, augmentant ainsi la transmission de la lumière, qui est détectée par une cellule photoélectrique.
Agrégométrie d'impédance sur sang total
L’agrégométrie d’impédance sur sang total (WBA) mesure la variation d’impédance électrique entre deux électrodes lorsque l’agrégation plaquettaire est induite par un agoniste. La lumiagrégométrie sur sang total peut accroître la sensibilité du test aux altérations de la sécrétion des granules plaquettaires.
PFA-100
Le PFA-100 (Test de Fonction Plaquettaire – 100) est un système d'analyse de la fonction plaquettaire. Du sang total citraté est aspiré à travers une cartouche jetable munie d'une ouverture dans une membrane recouverte soit de collagène et d'épinéphrine, soit de collagène et d'ADP. Ces agonistes induisent l'adhésion, l'activation et l'agrégation plaquettaires, entraînant une occlusion rapide de l'ouverture et l'arrêt du flux sanguin, appelé temps de fermeture (TC). Un TC élevé avec l'épinéphrine et le collagène peut indiquer des anomalies intrinsèques telles que la maladie de von Willebrand , l'urémie ou la présence d'inhibiteurs plaquettaires circulants. Un test de confirmation avec le collagène et l'ADP permet de déterminer si le TC anormal observé avec le collagène et l'épinéphrine est dû aux effets de l'acide acétylsulfosalicylique (aspirine) ou de médicaments contenant des inhibiteurs. Le PFA-100 est très sensible à la maladie de von Willebrand, mais sa sensibilité aux anomalies de la fonction plaquettaire est modérée.
Signification clinique
Des saignements spontanés et excessifs peuvent survenir en raison de troubles plaquettaires. Ces saignements peuvent être causés par un nombre insuffisant de plaquettes, des plaquettes dysfonctionnelles ou une densité plaquettaire supérieure à 1 million/microlitre. (Un nombre excessif de plaquettes entraîne un déficit relatif en facteur von Willebrand par séquestration.)
Les saignements dus à un trouble plaquettaire ou à un trouble de la coagulation peuvent être distingués par leurs caractéristiques et leur localisation. Les saignements plaquettaires comprennent les saignements d'une plaie, rapides et abondants, mais contrôlables par compression ; les saignements spontanés de la peau provoquant une tache violacée, nommée selon sa taille : pétéchies , purpura , ecchymoses ; les saignements des muqueuses, entraînant des saignements des gencives, des épistaxis et des saignements gastro-intestinaux ; les ménorragies ; et les hémorragies intrarétiniennes et intracrâniennes.
Un nombre excessif de plaquettes, et/ou une réaction des plaquettes normales à des anomalies de la paroi vasculaire, peuvent entraîner une thrombose veineuse et une thrombose artérielle . Les symptômes dépendent du site de la thrombose.
Troubles
Les troubles plaquettaires peuvent survenir en raison d'un nombre insuffisant ou excessif de plaquettes, ou d'un dysfonctionnement plaquettaire.
Une faible concentration de plaquettes est appelée thrombocytopénie et résulte soit d'une diminution de la production, soit d'une destruction accrue des plaquettes, soit de leur séquestration dans une autre partie de l'organisme. Une concentration élevée de plaquettes est appelée thrombocytose et peut être congénitale , réactionnelle (aux cytokines ) ou due à une production non régulée : par exemple, dans le cadre d'un syndrome myéloprolifératif ou de certaines autres hémopathies myéloïdes .
Les plaquettes normales peuvent réagir à une anomalie de la paroi vasculaire plutôt qu'à une hémorragie, entraînant une adhésion/activation plaquettaire inappropriée et une thrombose : la formation d'un caillot à l'intérieur d'un vaisseau intact. Ce type de thrombose survient par des mécanismes différents de ceux d'un caillot normal : extension de la fibrine dans la thrombose veineuse ; extension d'une plaque artérielle instable ou rompue, provoquant une thrombose artérielle ; et thrombose microcirculatoire. Un thrombus artériel peut obstruer partiellement le flux sanguin, provoquant une ischémie en aval , ou l'obstruer complètement, provoquant une nécrose tissulaire en aval .
| ADP | Épinéphrine | collagène | Ristorcétine | |
|---|---|---|---|---|
| Défaut du récepteur P2Y (y compris le clopidogrel ) | Diminué | Normale | Normale | Normale |
| Défaut des récepteurs adrénergiques | Normale | Diminué | Normale | Normale |
| Défaut du récepteur du collagène | Normale | Normale | Diminué ou absent | Normale |
| Normale | Normale | Normale | Diminué ou absent | |
| Diminué | Diminué | Diminué | Normal ou diminué | |
| Insuffisance du pool de stockage | Absence de deuxième vague | Partiel | ||
| Trouble lié à l'aspirine ou à un phénomène similaire à l'aspirine | Absence de deuxième vague | Absent | Normale | |
Thrombocytopénie
- Thrombocytopénie immunitaire (PTI) — anciennement connue sous le nom de purpura thrombocytopénique immunitaire et de purpura thrombocytopénique idiopathique
- Splénomégalie
- Thrombocytopénie familiale
- Chimiothérapie
- Babésiose
- la dengue
- Onyalai
- purpura thrombotique thrombocytopénique
- Syndrome HELLP
- syndrome hémolytique et urémique
- Purpura thrombopénique induit par les médicaments (cinq médicaments connus — le plus problématique est la thrombocytopénie induite par l'héparine (TIH))
- associée à la grossesse
- Allo-immunité néonatale associée
- Anémie aplastique
- associé à la transfusion
- Pseudothrombocytopénie
- Thrombocytopénie thrombotique immunologique induite par la vaccination (VITT)
Fonction plaquettaire altérée
- Congénital
- Troubles de l'adhérence
- Troubles de l'activation
- Troubles de la quantité ou de la libération des granules
- Syndrome d'Hermansky-Pudlak
- Syndrome des plaquettes grises
- défaut du récepteur ADP
- Activité de la cyclooxygénase diminuée
- Déficit du pool de stockage plaquettaire
- Troubles de l'agrégation
- Troubles de l'activité coagulante
- Acquis
- Troubles de l'adhérence
- hémoglobinurie paroxystique nocturne
- Asthme
- Maladie respiratoire exacerbée par l'aspirine (AERD/triade de Samter)
- Cancer
- Paludisme
- Activité de la cyclooxygénase diminuée
- Troubles de l'adhérence
Thrombocytose et thrombocythémie
- Réactif
- Infection chronique
- inflammation chronique
- Malignité
- Hyposplénisme (post-splénectomie)
- carence en fer
- Hémorragie aiguë
- Néoplasies myéloprolifératives — les plaquettes sont à la fois élevées et activées
- Associé à d'autres néoplasies myéloïdes
- Congénital
Pharmacologie
Médicaments anti-inflammatoires
Certains médicaments anti-inflammatoires ont pour effet indésirable de supprimer la fonction plaquettaire normale. Il s'agit des anti-inflammatoires non stéroïdiens (AINS). L'aspirine perturbe de façon irréversible la fonction plaquettaire en inhibant la cyclooxygénase -1 (COX-1), et donc l'hémostase normale. Les plaquettes ainsi altérées sont incapables de produire de la cyclooxygénase car elles sont dépourvues d'ADN. La fonction plaquettaire normale ne se rétablit qu'après l'arrêt de la prise d'aspirine et le remplacement d'un nombre suffisant de plaquettes affectées par de nouvelles, ce qui peut prendre plus d'une semaine. L'ibuprofène , un autre AINS , a un effet moins prolongé : la fonction plaquettaire se rétablit généralement en 24 heures , et la prise d'ibuprofène avant l'aspirine prévient les effets irréversibles de cette dernière
Médicaments qui suppriment la fonction plaquettaire
Ces médicaments sont utilisés pour prévenir la formation de thrombus.
Agents oraux
Agents intraveineux
Médicaments qui stimulent la production de plaquettes
Thérapies
Transfusion
Collection

Les plaquettes sont soit isolées à partir d'unités de sang total collectées et regroupées pour constituer une dose thérapeutique, soit collectées par aphérèse plaquettaire : le sang est prélevé chez le donneur, passe dans un appareil qui en sépare les plaquettes, puis le reste est réinjecté au donneur en circuit fermé. La norme du secteur exige que les plaquettes soient testées pour détecter la présence de bactéries avant la transfusion afin d'éviter les réactions septiques, qui peuvent être fatales. Récemment, les normes de l'AABB pour les banques de sang et les services de transfusion (5.1.5.1) ont autorisé l'utilisation de technologies de réduction des agents pathogènes comme alternative aux tests bactériens sur les plaquettes.
Les plaquettes issues de sang total poolé, parfois appelées plaquettes « aléatoires », sont séparées selon deux méthodes. Aux États-Unis, une unité de sang total est placée dans une grande centrifugeuse à vitesse réduite. Dans ces conditions, les plaquettes restent en suspension dans le plasma. Le plasma riche en plaquettes (PRP) est séparé des globules rouges, puis centrifugé à une vitesse plus élevée pour récupérer les plaquettes du plasma. Dans d'autres régions du monde, l'unité de sang total est centrifugée selon des paramètres qui permettent aux plaquettes de se retrouver en suspension dans la couche leucocytaire , qui contient les plaquettes et les globules blancs. La couche leucocytaire est isolée dans une poche stérile, mise en suspension dans une petite quantité de globules rouges et de plasma, puis centrifugée une seconde fois pour séparer les plaquettes et le plasma des globules rouges et blancs. Quelle que soit la méthode de préparation initiale, plusieurs dons peuvent être combinés dans un seul récipient à l'aide d'un dispositif de connexion stérile afin de fabriquer un produit unique à la dose thérapeutique souhaitée.
Les plaquettes d'aphérèse sont collectées à l'aide d'un appareil mécanique qui prélève le sang du donneur et le centrifuge afin de séparer les plaquettes et les autres composants à récupérer. Le sang restant est réinjecté au donneur. L'avantage de cette méthode est qu'un seul don permet d'obtenir au moins une dose thérapeutique, contrairement aux dons multiples nécessaires pour les plaquettes issues de sang total. Ainsi, le receveur est exposé à moins de donneurs et court moins de risques de maladies transmissibles par transfusion et d'autres complications. Parfois, une personne, comme un patient atteint de cancer nécessitant des transfusions régulières de plaquettes, reçoit des dons répétés d'un même donneur afin de minimiser les risques. La réduction des agents pathogènes dans les plaquettes, par exemple grâce à des traitements à la riboflavine et aux UV, permet de diminuer la charge infectieuse des agents pathogènes contenus dans les produits sanguins donnés. Un autre procédé de traitement photochimique utilisant l'amotosalène et les UVA a été mis au point pour l'inactivation des virus, des bactéries, des parasites et des leucocytes. De plus, les plaquettes d’aphérèse ont tendance à contenir moins de globules rouges contaminants car la méthode de collecte est plus efficace que la centrifugation « douce ».
Stockage
Les plaquettes collectées par l'une ou l'autre méthode ont une durée de conservation typique de cinq jours. Ceci entraîne des pénuries, car les dons destinés aux tests nécessitent souvent jusqu'à une journée entière. Aucune solution de conservation efficace n'a été mise au point pour les plaquettes.
Les plaquettes sont conservées sous agitation constante à une température une bactériémie . Aux États-Unis, les produits doivent être testés pour détecter toute contamination bactérienne avant transfusion.

plaquettes cryoconservées
Cryopreserved platelets are platelet components frozen for long-term storage, typically using dimethyl sulfoxide as a cryoprotectant. They have been investigated as an alternative to conventional liquid-stored platelets, which have a short shelf life of approximately 5 to 7 days, limiting availability and contributing to wastage. The longer shelf life of cryopreserved platelets may be useful in settings where maintaining an inventory of liquid-stored platelets is difficult, including small, rural, military, or resource-limited hospitals. The CLIP-II randomized clinical trial found that cryopreserved platelets did not meet the criterion for noninferiority compared with liquid-stored platelets for bleeding control in cardiac surgery, although no increase in prespecified adverse events was observed. A trial-based economic evaluation alongside CLIP-II found that cryopreserved platelets were more costly and less effective than liquid-stored platelets for treating active bleeding in patients undergoing cardiac surgery in metropolitan Australian hospitals.
Delivery
The change in the recipient's platelet count after transfusion is termed the "increment" and is calculated by subtracting the pre-transfusion platelet count from the post-transfusion count. Many factors affect the increment including body size, the number of platelets transfused, and clinical features that may cause premature destruction of the transfused platelets. When recipients fail to demonstrate an adequate post-transfusion increment, this is termed platelet transfusion refractoriness.
Platelets, either apheresis-derived or random-donor, can be processed through a volume reduction process. In this process, the platelets are spun in a centrifuge and plasma is removed, leaving 10 to 100 mL of platelet concentrate. Such volume-reduced platelets are normally transfused only to neonatal and pediatric patients when a large volume of plasma could overload the child's small circulatory system. The lower volume of plasma also reduces the chances of an adverse transfusion reaction to plasma proteins. Volume reduced platelets have a shelf life of four hours.
Wound repair
Platelets release platelet-derived growth factor (PDGF), a potent chemotactic agent; and TGF beta, which stimulates the deposition of extracellular matrix; fibroblast growth factor, insulin-like growth factor 1, platelet-derived epidermal growth factor, and vascular endothelial growth factor. Local application of these factors in increased concentrations through platelet-rich plasma (PRP) is used as an adjunct in wound healing.
Non-mammals
Instead of platelets, non-mammalian vertebrates have nucleated thrombocytes, which resemble B lymphocytes in morphology. They aggregate in response to thrombin, but not to ADP, serotonin, nor adrenaline, as platelets do.
History
- George Gulliver in 1841 drew pictures of platelets using the twin lens (compound) microscope invented in 1830 by Joseph Jackson Lister. This microscope improved resolution sufficiently to make it possible to see platelets for the first time.
- William Addison in 1842 drew pictures of a platelet-fibrin clot.
- Lionel Beale in 1864 was the first to publish a drawing showing platelets.
- Max Schultze a décrit en 1865 ce qu'il appelait des « sphérules », qu'il a noté être beaucoup plus petites que les globules rouges, parfois agglutinées, et se trouvaient parfois dans des collections de matériel fibrineux.
- En 1882, Giulio Bizzozero étudia au microscope in vivo le sang des amphibiens . Il nomma les sphérules de Schultze (en italien) piastrine : petites plaques. Bizzozero proposa peut-être le nom Blutplattchen.
- William Osler observa les plaquettes et, dans des conférences publiées en 1886, les appela un troisième corpuscule et une plaque sanguine ; et les décrivit comme « un disque protoplasmique incolore ».
- James Wright a examiné des frottis sanguins en utilisant la coloration qui porte son nom, et a utilisé le terme « plaquettes » dans sa publication de 1906, passant à « plaquettes » dans sa publication de 1910.