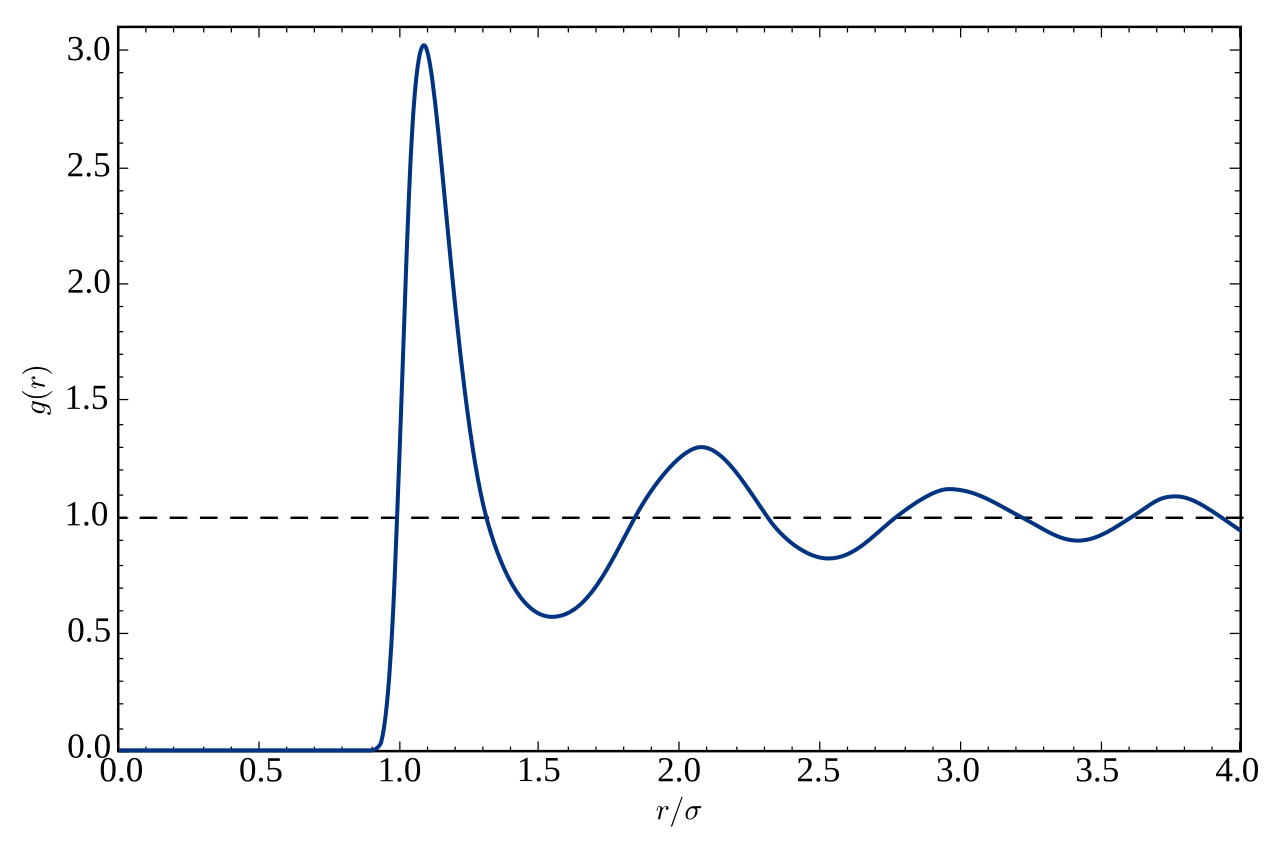
En mécanique statistique , la fonction de distribution radiale (ou fonction de corrélation de paires ) dans un système de particules (atomes, molécules, colloïdes, etc.), décrit comment la densité varie en fonction de la distance par rapport à une particule de référence.
Si une particule donnée est considérée comme étant à l'origine O , et si est la densité numérique moyenne des particules, alors la densité locale moyenne temporelle à une certaine distance de O est . Cette définition simplifiée est valable pour un système homogène et isotrope . Un cas plus général sera considéré ci-dessous.
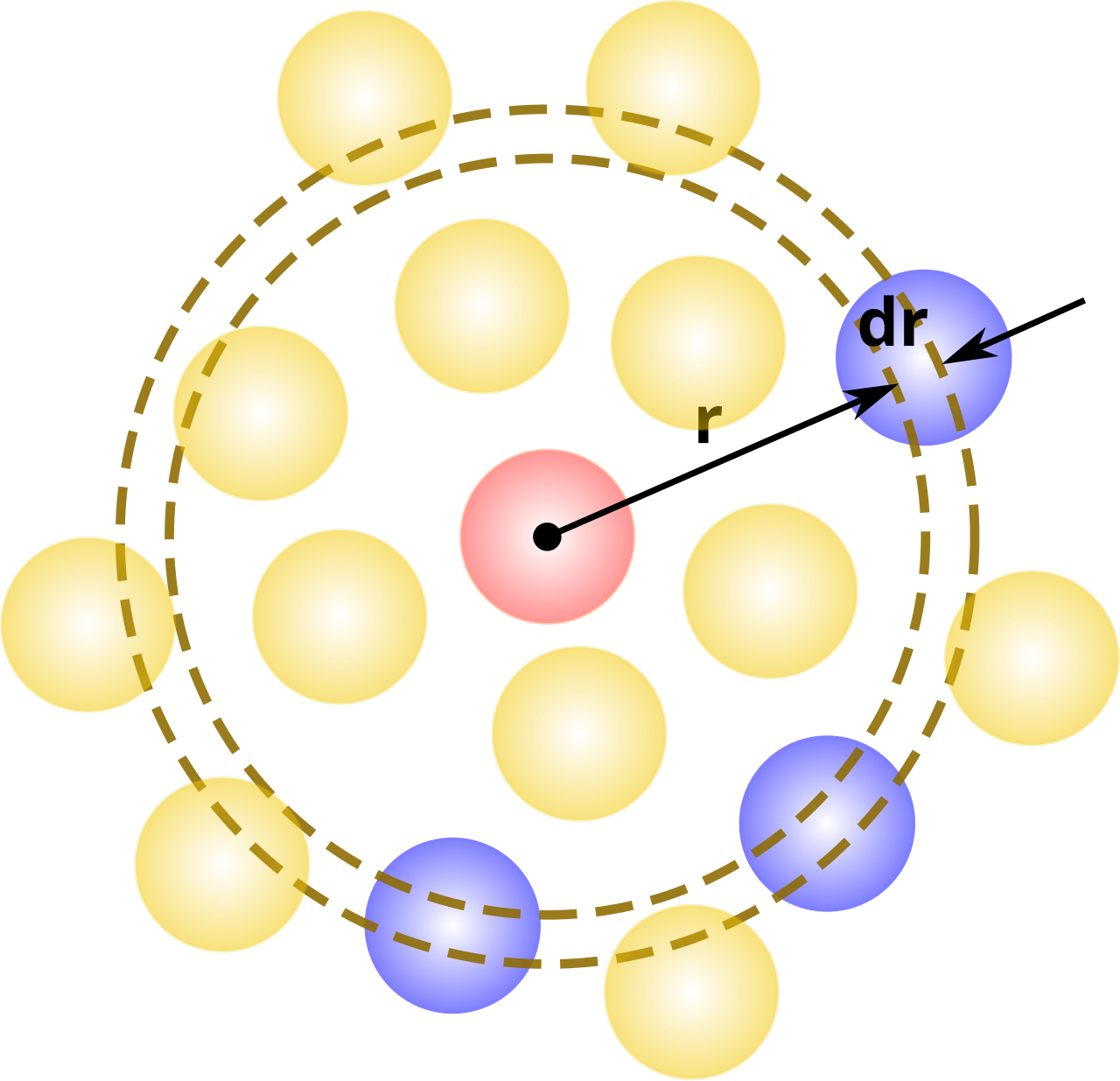
En termes simples, il s'agit d'une mesure de la probabilité de trouver une particule à une distance d'une particule de référence donnée, par rapport à celle d'un gaz idéal. L'algorithme général consiste à déterminer combien de particules se trouvent à une distance et à une distance d'une particule. Ce thème général est représenté à droite, où la particule rouge est notre particule de référence, et les particules bleues sont celles dont les centres se trouvent à l'intérieur de la coque circulaire, pointillée en orange.
La fonction de distribution radiale est généralement déterminée en calculant la distance entre toutes les paires de particules et en les regroupant dans un histogramme. L'histogramme est ensuite normalisé par rapport à un gaz idéal, où les histogrammes de particules sont complètement décorrélés. Pour trois dimensions, cette normalisation est la densité numérique du système multipliée par le volume de la coque sphérique, qui peut symboliquement être exprimée comme .
Étant donnée une fonction d'énergie potentielle , la fonction de distribution radiale peut être calculée soit par des méthodes de simulation informatique comme la méthode de Monte Carlo , soit par l' équation d'Ornstein-Zernike , en utilisant des relations de fermeture approximatives comme l' approximation de Percus-Yevick ou la théorie de la chaîne hypernettée . Elle peut également être déterminée expérimentalement, par des techniques de diffusion du rayonnement ou par visualisation directe pour des particules suffisamment grandes (de la taille d'un micromètre) via la microscopie traditionnelle ou confocale.
La fonction de distribution radiale est d'une importance fondamentale car elle peut être utilisée, en utilisant la théorie de la solution de Kirkwood-Buff , pour relier les détails microscopiques aux propriétés macroscopiques. De plus, grâce à l'inversion de la théorie de Kirkwood-Buff, il est possible d'obtenir les détails microscopiques de la fonction de distribution radiale à partir des propriétés macroscopiques. La fonction de distribution radiale peut également être inversée pour prédire la fonction d'énergie potentielle en utilisant l' équation d'Ornstein-Zernike ou le raffinement du potentiel optimisé par la structure.
Définition
Considérons un système de particules dans un volume (pour une densité numérique moyenne ) et à une température (définissons aussi ; est la constante de Boltzmann). Les coordonnées des particules sont , avec . L' énergie potentielle due à l'interaction entre particules est et nous ne considérons pas le cas d'un champ appliqué de l'extérieur.
Les moyennes appropriées sont prises dans l' ensemble canonique , avec l'intégrale de configuration, prise sur toutes les combinaisons possibles de positions de particules. La probabilité d'une configuration élémentaire, à savoir de trouver la particule 1 dans , la particule 2 dans , etc. est donnée par
Le nombre total de particules est énorme, ce qui en soi n'est pas très utile. Cependant, on peut aussi obtenir la probabilité d'une configuration réduite, où les positions des seules particules sont fixées, dans , sans aucune contrainte sur les particules restantes. Pour cela, il faut intégrer ( 1 ) sur les coordonnées restantes :
Si les particules ne sont pas en interaction, dans le sens où l'énergie potentielle de chaque particule ne dépend d'aucune des autres particules, alors la fonction de partition se factorise et la probabilité d'une configuration élémentaire se décompose avec des arguments indépendants en un produit de probabilités de particules individuelles,
Notez que pour les particules non interactives, la probabilité est symétrique dans ses arguments. Ce n'est pas vrai en général, et l'ordre dans lequel les positions occupent les emplacements d'argument de est important. Étant donné un ensemble de positions, la manière dont les particules peuvent occuper ces positions est La probabilité que ces positions SONT occupées est trouvée en additionnant toutes les configurations dans lesquelles une particule est à chacun de ces emplacements. Cela peut être fait en prenant chaque permutation , , dans le groupe symétrique sur les objets, , pour écrire . Pour moins de positions, nous intégrons sur les arguments étrangers, et incluons un facteur de correction pour éviter le surcomptage, Cette quantité est appelée la fonction de densité à n particules . Pour des particules indiscernables , on pourrait permuter toutes les positions de particules, , sans changer la probabilité d'une configuration élémentaire, , de sorte que la fonction de densité à n particules se réduise à L'intégration de la densité à n particules donne le facteur de permutation , en comptant le nombre de façons dont on peut sélectionner séquentiellement des particules à placer aux positions parmi le total des particules. Voyons maintenant comment interpréter ces fonctions pour différentes valeurs de .
Pour , nous avons la densité d'une particule. Pour un cristal, c'est une fonction périodique avec des maxima nets aux sites du réseau. Pour un gaz sans interaction, elle est indépendante de la position et égale à la densité numérique globale, , du système. Pour voir cela, notez d'abord que dans le volume occupé par le gaz, et 0 partout ailleurs. La fonction de partition dans ce cas est
à partir de laquelle la définition donne le résultat souhaité
En fait, pour ce cas particulier, chaque densité de n particules est indépendante des coordonnées et peut être calculée explicitement. Pour , la densité de n particules non interactives est approximativement de . Avec cela en main, la fonction de corrélation à n points est définie en factorisant la contribution non interactive , Explicitement, cette définition se lit là où il est clair que la fonction de corrélation à n points est sans dimension.
Relations impliquantg(l)
Facteur de structure
La fonction de corrélation du second ordre revêt une importance particulière, car elle est directement liée (via une transformée de Fourier ) au facteur de structure du système et peut donc être déterminée expérimentalement à l'aide de la diffraction des rayons X ou de la diffraction des neutrons .
Si le système est constitué de particules sphériquement symétriques, ne dépend que de la distance relative entre elles, . Nous abandonnerons l'indice et l'exposant : . En prenant la particule 0 comme fixée à l'origine des coordonnées, est le nombre moyen de particules (parmi les ) qui se trouvent dans le volume autour de la position .
Nous pouvons formellement compter ces particules et prendre la moyenne via l'expression , avec la moyenne d'ensemble, ce qui donne :
où la seconde égalité requiert l'équivalence des particules . La formule ci-dessus est utile pour se rapporter au facteur de structure statique , défini par , puisque nous avons :
et donc :
Cette équation n'est valable que dans le sens des distributions , car n'est pas normalisée : , de sorte que diverge comme le volume , conduisant à un pic de Dirac à l'origine pour le facteur de structure. Comme cette contribution est inaccessible expérimentalement, nous pouvons la soustraire de l'équation ci-dessus et redéfinir le facteur de structure comme une fonction régulière :
Enfin, on renomme et, si le système est liquide, on peut invoquer son isotropie :
Équation de compressibilité
En évaluant ( 6 ) et en utilisant la relation entre la compressibilité isotherme et le facteur de structure à l'origine, on obtient l' équation de compressibilité :
Potentiel de force moyenne
Il peut être démontré que la fonction de distribution radiale est liée au potentiel à deux particules de la force moyenne par :
Dans la limite diluée, le potentiel de force moyenne est le potentiel de paire exact sous lequel la configuration du point d'équilibre a une valeur donnée .
Équation énergétique
Si les particules interagissent via des potentiels identiques par paires : , l'énergie interne moyenne par particule est : 
Équation d'état de pression
Le développement de l’ équation du viriel donne l’équation d’état de pression :
Propriétés thermodynamiques en 3D
La fonction de distribution radiale est une mesure importante car plusieurs propriétés thermodynamiques clés, telles que l'énergie potentielle et la pression, peuvent être calculées à partir de celle-ci.
Pour un système 3D où les particules interagissent via des potentiels par paires, l'énergie potentielle du système peut être calculée comme suit :
où N est le nombre de particules dans le système, est la densité numérique, est le potentiel de paire .
La pression du système peut également être calculée en reliant le 2e coefficient du viriel à . La pression peut être calculée comme suit :
Notez que les résultats de l'énergie potentielle et de la pression ne seront pas aussi précis que le calcul direct de ces propriétés en raison de la moyenne impliquée dans le calcul de .
Approximations
Pour les systèmes dilués (par exemple les gaz), les corrélations dans les positions des particules prises en compte ne sont dues qu'au potentiel engendré par la particule de référence, en négligeant les effets indirects. En première approximation, elle est donc simplement donnée par la loi de distribution de Boltzmann :
Si étaient nuls pour tous – c'est-à-dire si les particules n'exerçaient aucune influence les unes sur les autres, alors pour tous et la densité locale moyenne serait égale à la densité moyenne : la présence d'une particule en O n'influencerait pas la distribution des particules autour d'elle et le gaz serait idéal. Pour des distances telles que est significative, la densité locale moyenne sera différente de la densité moyenne , selon le signe de (plus élevée pour une énergie d'interaction négative et plus faible pour une énergie positive ).
Au fur et à mesure que la densité du gaz augmente, la limite de faible densité devient de moins en moins précise puisqu'une particule située en subit non seulement l'interaction avec la particule en O mais aussi avec les autres voisines, elles-mêmes influencées par la particule de référence. Cette interaction médiatisée augmente avec la densité, puisqu'il y a davantage de voisins avec lesquels interagir : il est logique d'écrire un développement de densité de , qui ressemble à l' équation du viriel :
Cette similitude n'est pas fortuite ; en effet, en remplaçant ( 12 ) dans les relations ci-dessus les paramètres thermodynamiques (équations 7 , 9 et 10 ) on obtient les développements viriels correspondants. La fonction auxiliaire est connue sous le nom de fonction de distribution de cavité . Il a été démontré que pour les fluides classiques à densité fixe et à température positive fixe, le potentiel de paire effectif qui génère une donnée sous équilibre est unique à une constante additive près, si elle existe.
Ces dernières années, une certaine attention a été accordée au développement de fonctions de corrélation de paires pour des données spatialement discrètes telles que des treillis ou des réseaux.
Expérimental
On peut déterminer indirectement (via sa relation avec le facteur de structure ) en utilisant des données de diffusion de neutrons ou de diffusion de rayons X. La technique peut être utilisée à des échelles de longueur très courtes (jusqu'au niveau atomique ) mais implique un moyennage spatial et temporel important (sur la taille de l'échantillon et le temps d'acquisition, respectivement). De cette manière, la fonction de distribution radiale a été déterminée pour une grande variété de systèmes, allant des métaux liquides aux colloïdes chargés. Passer de l'expérimental à l'expérimental n'est pas simple et l'analyse peut être assez complexe.
Il est également possible de calculer directement en extrayant les positions des particules à partir de la microscopie traditionnelle ou confocale. Cette technique est limitée aux particules suffisamment grosses pour la détection optique (de l'ordre du micromètre), mais elle présente l'avantage d'être résolue dans le temps de sorte que, outre les informations statiques, elle donne également accès à des paramètres dynamiques (par exemple les constantes de diffusion ) et également résolue dans l'espace (au niveau de la particule individuelle), ce qui lui permet de révéler la morphologie et la dynamique des structures locales dans les cristaux colloïdaux, les verres, les gels, et les interactions hydrodynamiques.
La visualisation directe d'une fonction de corrélation de paires complète (dépendante de la distance et de l'angle) a été obtenue par microscopie à effet tunnel dans le cas de gaz moléculaires 2D.
Fonctions de corrélation d'ordre supérieur
Il a été noté que les fonctions de distribution radiale seules ne suffisent pas à caractériser les informations structurelles. Des processus ponctuels distincts peuvent posséder des fonctions de distribution radiale identiques ou pratiquement indiscernables, ce que l'on appelle le problème de dégénérescence. Dans de tels cas, des fonctions de corrélation d'ordre supérieur sont nécessaires pour décrire plus en détail la structure.
Les fonctions de distribution d'ordre supérieur ont été moins étudiées , car elles sont généralement moins importantes pour la thermodynamique du système ; en même temps, elles ne sont pas accessibles par les techniques de diffusion conventionnelles. Elles peuvent cependant être mesurées par diffusion cohérente des rayons X et sont intéressantes dans la mesure où elles peuvent révéler des symétries locales dans des systèmes désordonnés.