L' algorithme de Smith-Waterman effectue un alignement de séquences locales , c'est-à-dire qu'il permet de déterminer les régions similaires entre deux chaînes de séquences d'acides nucléiques ou de séquences protéiques . Au lieu d'examiner la séquence entière , l'algorithme de Smith-Waterman compare des segments de toutes les longueurs possibles et optimise la mesure de similarité .
L'algorithme a été proposé pour la première fois par Temple F. Smith et Michael S. Waterman en 1981. Comme l' algorithme Needleman-Wunsch , dont il est une variante, Smith-Waterman est un algorithme de programmation dynamique . En tant que tel, il a la propriété souhaitable de garantir la recherche de l'alignement local optimal par rapport au système de notation utilisé (qui comprend la matrice de substitution et le système de notation des écarts ). La principale différence avec l' algorithme Needleman-Wunsch est que les cellules de la matrice de notation négative sont définies à zéro. La procédure de traçage commence à la cellule de la matrice de notation la plus élevée et se poursuit jusqu'à ce qu'une cellule avec un score zéro soit rencontrée, ce qui donne l'alignement local de notation le plus élevé. En raison de sa complexité temporelle cubique, il ne peut souvent pas être appliqué de manière pratique à des problèmes à grande échelle et est remplacé par des alternatives plus efficaces sur le plan informatique telles que (Gotoh, 1982), ( Altschul et Erickson, 1986), et (Myers et Miller, 1988).
Histoire
En 1970, Saul B. Needleman et Christian D. Wunsch ont proposé un algorithme d'homologie heuristique pour l'alignement des séquences, également appelé algorithme Needleman-Wunsch. Il s'agit d'un algorithme d'alignement global qui nécessite des étapes de calcul ( et sont les longueurs des deux séquences alignées). Il utilise le calcul itératif d'une matrice dans le but de montrer l'alignement global. Au cours de la décennie suivante, Sankoff, Reichert, Beyer et d'autres ont formulé des algorithmes heuristiques alternatifs pour analyser les séquences de gènes. Sellers a introduit un système de mesure des distances de séquence. En 1976, Waterman et al. ont ajouté le concept d'écarts dans le système de mesure d'origine. En 1981, Smith et Waterman ont publié leur algorithme Smith-Waterman pour calculer l'alignement local.
L'algorithme de Smith-Waterman est assez exigeant en termes de temps : pour aligner deux séquences de longueurs et , il faut du temps. Gotoh et Altschul ont optimisé l'algorithme en étapes. La complexité spatiale a été optimisée par Myers et Miller de à (linéaire), où est la longueur de la séquence la plus courte, pour le cas où un seul des nombreux alignements optimaux possibles est souhaité. Chowdhury, Le et Ramachandran ont ensuite optimisé les performances du cache de l'algorithme tout en gardant l'utilisation de l'espace linéaire dans la longueur totale des séquences d'entrée.
Motivation
Ces dernières années, les projets de génomique menés sur divers organismes ont généré des quantités massives de données de séquences de gènes et de protéines, ce qui nécessite une analyse informatique. L'alignement des séquences montre les relations entre les gènes ou entre les protéines, ce qui conduit à une meilleure compréhension de leur homologie et de leur fonctionnalité. L'alignement des séquences peut également révéler des domaines et des motifs conservés .
L'alignement local est motivé par la difficulté d'obtenir des alignements corrects dans des régions de faible similarité entre des séquences biologiques éloignées, car les mutations ont ajouté trop de « bruit » au cours de l'évolution pour permettre une comparaison significative de ces régions. L'alignement local évite complètement ces régions et se concentre sur celles qui ont un score positif, c'est-à-dire celles qui ont un signal de similarité conservé au cours de l'évolution. Une condition préalable à l'alignement local est un score d'espérance négatif. Le score d'espérance est défini comme le score moyen que le système de notation ( matrice de substitution et pénalités d'écart ) produirait pour une séquence aléatoire.
Une autre motivation pour l'utilisation des alignements locaux est qu'il existe un modèle statistique fiable (développé par Karlin et Altschul) pour les alignements locaux optimaux. L'alignement de séquences non apparentées tend à produire des scores d'alignement local optimaux qui suivent une distribution de valeurs extrêmes. Cette propriété permet aux programmes de produire une valeur d'espérance pour l'alignement local optimal de deux séquences, qui est une mesure de la fréquence à laquelle deux séquences non apparentées produiraient un alignement local optimal dont le score est supérieur ou égal au score observé. Des valeurs d'espérance très faibles indiquent que les deux séquences en question pourraient être homologues , ce qui signifie qu'elles pourraient partager un ancêtre commun.
Algorithme
Soient et les séquences à aligner, où et sont respectivement les longueurs de et .
- Déterminer la matrice de substitution et le schéma de pénalité d'écart.
- Construisez une matrice de notation et initialisez sa première ligne et sa première colonne. La taille de la matrice de notation est . La matrice utilise une indexation basée sur 0.
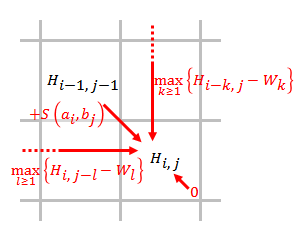
- Remplissez la matrice de notation en utilisant l’équation ci-dessous.
- où
- Traceback. En commençant par le score le plus élevé dans la matrice de notation et en terminant par une cellule de la matrice qui a un score de 0, traceback basé sur la source de chaque score de manière récursive pour générer le meilleur alignement local.
Explication
L'algorithme de Smith-Waterman aligne deux séquences par correspondances/non-correspondances (également appelées substitutions), insertions et suppressions. Les insertions et les suppressions sont les opérations qui introduisent des espaces, représentés par des tirets. L'algorithme de Smith-Waterman comporte plusieurs étapes :
- Déterminer la matrice de substitution et le système de pénalité d'écart . Une matrice de substitution attribue à chaque paire de bases ou d'acides aminés un score pour la correspondance ou la non-correspondance. En général, les correspondances obtiennent des scores positifs, tandis que les non-correspondances obtiennent des scores relativement plus faibles. Une fonction de pénalité d'écart détermine le coût du score pour l'ouverture ou l'extension des écarts. Il est suggéré aux utilisateurs de choisir le système de notation approprié en fonction des objectifs. En outre, il est également recommandé d'essayer différentes combinaisons de matrices de substitution et de pénalités d'écart.
- Initialisez la matrice de notation . Les dimensions de la matrice de notation sont respectivement 1+longueur de chaque séquence. Tous les éléments de la première ligne et de la première colonne sont définis à 0. La première ligne et la première colonne supplémentaires permettent d'aligner une séquence sur une autre à n'importe quelle position, et leur définition à 0 permet d'éliminer toute pénalité pour l'espace terminal.
- Notation . Notez chaque élément de gauche à droite, de haut en bas dans la matrice, en tenant compte des résultats des substitutions (scores diagonaux) ou de l'ajout d'écarts (scores horizontaux et verticaux). Si aucun des scores n'est positif, cet élément obtient un 0. Sinon, le score le plus élevé est utilisé et la source de ce score est enregistrée.
- Traceback . En commençant par l'élément avec le score le plus élevé, traceback basé sur la source de chaque score de manière récursive, jusqu'à ce que 0 soit rencontré. Les segments qui ont le score de similarité le plus élevé basé sur le système de notation donné sont générés dans ce processus. Pour obtenir le deuxième meilleur alignement local, appliquez le processus de traceback en commençant par le deuxième score le plus élevé en dehors de la trace du meilleur alignement.
Comparaison avec l'algorithme Needleman-Wunsch
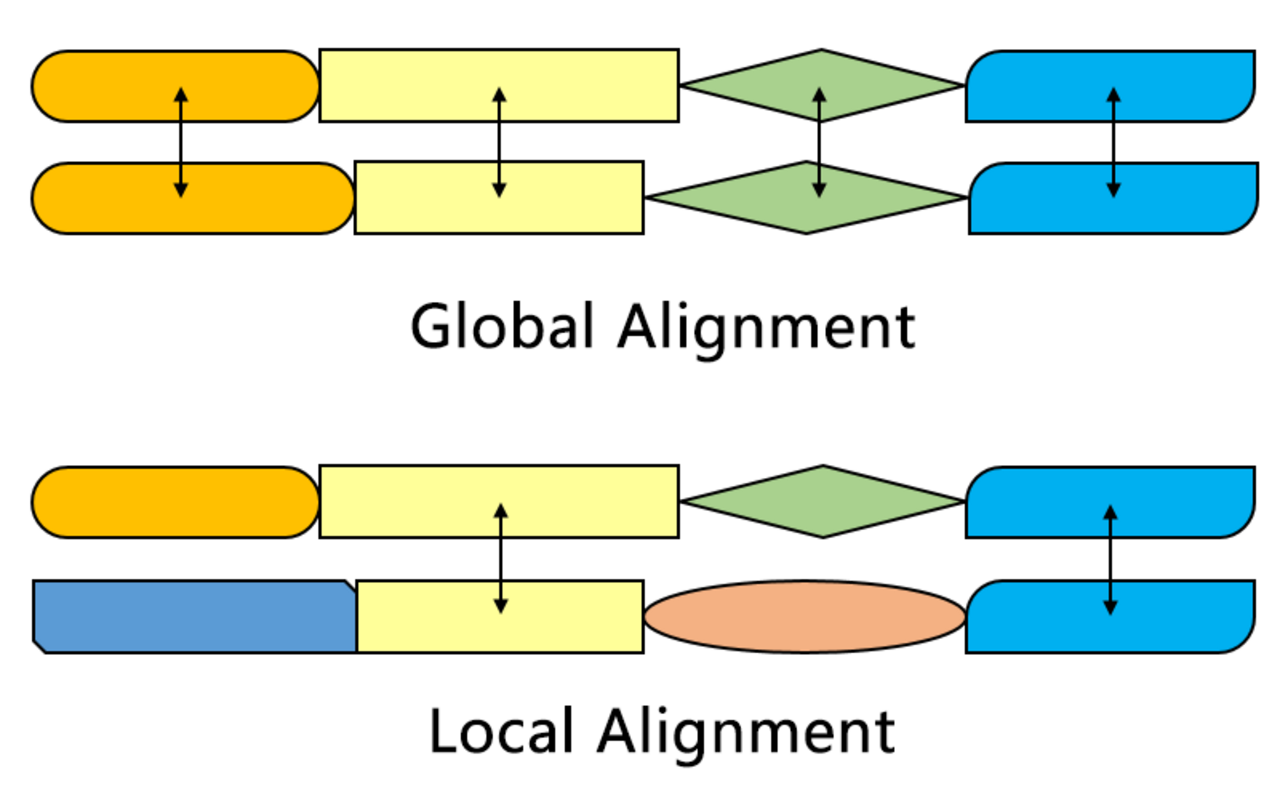
L'algorithme de Smith-Waterman recherche les segments de deux séquences qui présentent des similitudes, tandis que l'algorithme de Needleman-Wunsch aligne deux séquences complètes. Par conséquent, ils servent des objectifs différents. Les deux algorithmes utilisent les concepts d'une matrice de substitution, d'une fonction de pénalité d'écart, d'une matrice de notation et d'un processus de traçage. Les trois principales différences sont les suivantes :
L'une des distinctions les plus importantes est qu'aucun score négatif n'est attribué dans le système de notation de l'algorithme Smith-Waterman, ce qui permet un alignement local. Lorsqu'un élément a un score inférieur à zéro, cela signifie que les séquences jusqu'à cette position n'ont aucune similitude ; cet élément sera alors mis à zéro pour éliminer l'influence de l'alignement précédent. De cette façon, le calcul peut continuer à trouver l'alignement dans n'importe quelle position par la suite.
La matrice de notation initiale de l'algorithme de Smith-Waterman permet d'aligner n'importe quel segment d'une séquence sur une position arbitraire de l'autre séquence. Cependant, dans l'algorithme de Needleman-Wunsch, la pénalité d'écart de fin doit également être prise en compte afin d'aligner les séquences complètes.
Matrice de substitution
Chaque substitution de base ou d'acide aminé est associée à un score. En général, les correspondances se voient attribuer des scores positifs et les mésappariements des scores relativement plus faibles. Prenons l'exemple d'une séquence d'ADN. Si les correspondances obtiennent +1, les mésappariements -1, alors la matrice de substitution est la suivante :
Cette matrice de substitution peut être décrite comme :
Différentes substitutions de bases ou d'acides aminés peuvent avoir des scores différents. La matrice de substitution des acides aminés est généralement plus compliquée que celle des bases. Voir PAM , BLOSUM .
Pénalité d'écart
La pénalité d'écart désigne les scores d'insertion ou de suppression. Une stratégie simple de pénalité d'écart consiste à utiliser un score fixe pour chaque écart. En biologie, cependant, le score doit être compté différemment pour des raisons pratiques. D'une part, la similarité partielle entre deux séquences est un phénomène courant ; d'autre part, un seul événement de mutation génétique peut entraîner l'insertion d'un seul long écart. Par conséquent, les écarts connectés formant un long écart sont généralement plus favorisés que plusieurs écarts courts et dispersés. Afin de prendre en compte cette différence, les concepts d'ouverture d'écart et d'extension d'écart ont été ajoutés au système de notation. Le score d'ouverture d'écart est généralement plus élevé que le score d'extension d'écart. Par exemple, les paramètres par défaut dans EMBOSS Water sont : ouverture d'écart = 10, extension d'écart = 0,5.
Nous discutons ici de deux stratégies courantes pour la pénalité d'écart. Voir Pénalité d'écart pour plus de stratégies. Soit la fonction de pénalité d'écart pour un écart de longueur :
Linéaire
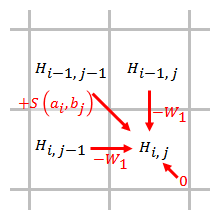
Une pénalité d'écart linéaire a les mêmes scores pour l'ouverture et l'extension d'un écart :
où est le coût d'un seul écart.
La pénalité d'écart est directement proportionnelle à la longueur de l'écart. Lorsque la pénalité d'écart linéaire est utilisée, l'algorithme de Smith-Waterman peut être simplifié comme suit :
L'algorithme simplifié utilise des étapes. Lorsqu'un élément est noté, seules les pénalités d'écart des éléments directement adjacents à cet élément doivent être prises en compte.
Affine
Une pénalité d'écart affine considère l'ouverture et l'extension de l'écart séparément :

où est la pénalité d'ouverture de l'écart, et est la pénalité d'extension de l'écart. Par exemple, la pénalité pour un écart de longueur 2 est .
Une pénalité d'écart arbitraire a été utilisée dans l'article original sur l'algorithme de Smith-Waterman. Il utilise des étapes, il est donc assez exigeant en temps. Gotoh a optimisé les étapes pour une pénalité d'écart affine à , mais l'algorithme optimisé ne tente de trouver qu'un seul alignement optimal, et l'alignement optimal n'est pas garanti d'être trouvé. Altschul a modifié l'algorithme de Gotoh pour trouver tous les alignements optimaux tout en maintenant la complexité de calcul. Plus tard, Myers et Miller ont souligné que l'algorithme de Gotoh et Altschul peut être encore modifié sur la base de la méthode publiée par Hirschberg en 1975, et ont appliqué cette méthode. L'algorithme de Myers et Miller peut aligner deux séquences en utilisant l'espace, étant la longueur de la séquence la plus courte. Chowdhury, Le et Ramachandran ont montré plus tard comment exécuter l'algorithme de Gotoh de manière efficace en termes de cache dans l'espace linéaire en utilisant une stratégie de division et de conquête récursive différente de celle utilisée par Hirschberg. L'algorithme résultant fonctionne plus rapidement que l'algorithme de Myers et Miller dans la pratique en raison de ses performances de cache supérieures.
Exemple de pénalité d'écart
Prenons l'exemple de l'alignement des séquences TACGGGCCCGCTAC et TAGCCCTATCGGTCA . Lorsque la fonction de pénalité d'écart linéaire est utilisée, le résultat est (Alignements effectués par EMBOSS Water. La matrice de substitution est DNAfull (score de similarité : +5 pour les caractères correspondants sinon -4). L'ouverture et l'extension de l'écart sont respectivement de 0,0 et 1,0) :
TACGGGCCCGCTA-C TA---G-CC-CTATC
Lorsque la pénalité d’écart affine est utilisée, le résultat est (l’ouverture et l’extension de l’écart sont respectivement de 5,0 et 1,0) :
TACGGGCCCGCTA TA---GCC--CTA
Cet exemple montre qu'une pénalité d'écart affine peut aider à éviter les petits écarts dispersés.
Matrice de notation
La fonction de la matrice de notation est d'effectuer des comparaisons un à un entre tous les composants de deux séquences et d'enregistrer les résultats d'alignement optimal. Le processus de notation reflète le concept de programmation dynamique. L'alignement optimal final est trouvé en développant de manière itérative l'alignement optimal croissant. En d'autres termes, l'alignement optimal actuel est généré en décidant quel chemin (correspondance/non-correspondance ou insertion d'un écart) donne le score le plus élevé par rapport à l'alignement optimal précédent. La taille de la matrice est la longueur d'une séquence plus 1 par la longueur de l'autre séquence plus 1. La première ligne et la première colonne supplémentaires servent à aligner une séquence sur n'importe quelle position de l'autre séquence. La première ligne et la première colonne sont toutes deux définies sur 0 afin que l'écart final ne soit pas pénalisé. La matrice de notation initiale est la suivante :
Exemple
Prenons comme exemple l'alignement des séquences d'ADN TGTTACGG et GGTTGACTA . Utilisez le schéma suivant :
- Matrice de substitution :
- Pénalité d'écart : (une pénalité d'écart linéaire de )
Initialisez et remplissez la matrice de notation, comme illustré ci-dessous. Cette figure montre le processus de notation des trois premiers éléments. La couleur jaune indique les bases prises en compte. La couleur rouge indique le score le plus élevé possible pour la cellule notée.
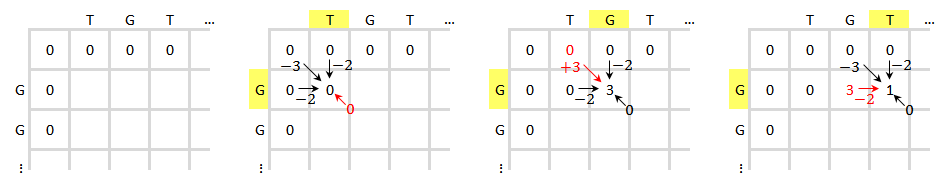
La matrice de notation terminée est présentée ci-dessous à gauche. La couleur bleue indique le score le plus élevé. Un élément peut recevoir un score de plusieurs éléments, chacun formera un chemin différent si cet élément est retracé. En cas de plusieurs scores les plus élevés, le traçage doit être effectué en commençant par chaque score le plus élevé. Le processus de traçage est présenté ci-dessous à droite. Le meilleur alignement local est généré dans le sens inverse.
Le résultat de l'alignement est :
GTT-AC GTTGAC
Mise en œuvre
Une implémentation de l'algorithme Smith-Waterman, SSEARCH, est disponible dans le package d'analyse de séquences FASTA de UVA FASTA Downloads. Cette implémentation comprend un code accéléré Altivec pour les processeurs PowerPC G4 et G5 qui accélère les comparaisons de 10 à 20 fois, en utilisant une modification de l'approche de Wozniak, 1997, et une vectorisation SSE2 développée par Farrar ce qui rend les recherches optimales dans les bases de données de séquences protéiques très pratiques. Une bibliothèque, SSW, étend l'implémentation de Farrar pour renvoyer des informations d'alignement en plus du score Smith-Waterman optimal.
Versions accélérées
FPGA
Cray a démontré l'accélération de l'algorithme Smith-Waterman à l'aide d'une plate-forme informatique reconfigurable basée sur des puces FPGA , avec des résultats montrant une accélération jusqu'à 28 fois supérieure à celle des solutions standard basées sur des microprocesseurs. Une autre version basée sur FPGA de l'algorithme Smith-Waterman montre des accélérations FPGA (Virtex-4) jusqu'à 100 fois sur un processeur Opteron à 2,2 GHz. Les systèmes TimeLogic DeCypher et CodeQuest accélèrent également Smith-Waterman et Framesearch à l'aide de cartes FPGA PCIe.
Une thèse de maîtrise de 2011 comprend une analyse de l'accélération Smith-Waterman basée sur FPGA.
Dans une publication de 2016, OpenCL code compiled with Xilinx SDAccel accelerates genome sequencing, beats CPU/GPU performance/W by 12-21x, une implémentation très efficace a été présentée. En utilisant une carte FPGA PCIe équipée d'un FPGA Xilinx Virtex-7 2000T, les performances par niveau de watt étaient supérieures à celles du CPU/GPU de 12 à 21 fois.
GPU
Le laboratoire national Lawrence Livermore et le Joint Genome Institute du département américain de l'énergie ont mis en œuvre une version accélérée des recherches d'alignement de séquences locales Smith-Waterman à l'aide d'unités de traitement graphique (GPU) avec des résultats préliminaires montrant une accélération de 2x par rapport aux implémentations logicielles. Une méthode similaire a déjà été mise en œuvre dans le logiciel Biofacet depuis 1997, avec le même facteur d'accélération.
Plusieurs implémentations GPU de l'algorithme sur la plateforme CUDA C de NVIDIA sont également disponibles. Comparés à l'implémentation CPU la plus connue (utilisant des instructions SIMD sur l'architecture x86), par Farrar, les tests de performances de cette solution utilisant une seule carte NVidia GeForce 8800 GTX montrent une légère augmentation des performances pour les séquences plus petites, mais une légère diminution des performances pour les plus grandes. Cependant, les mêmes tests exécutés sur deux cartes NVidia GeForce 8800 GTX sont presque deux fois plus rapides que l'implémentation Farrar pour toutes les tailles de séquence testées.
Une nouvelle implémentation GPU CUDA de SW est désormais disponible. Elle est plus rapide que les versions précédentes et supprime également les limitations sur la longueur des requêtes. Voir CUDASW++.
Onze implémentations SW différentes sur CUDA ont été signalées, dont trois signalent des accélérations de 30X.
Enfin, d'autres implémentations accélérées par GPU du Smith-Waterman peuvent être trouvées dans NVIDIA Parabricks , la suite logicielle de NVIDIA pour l'analyse du génome.
SIMD
En 2000, une implémentation rapide de l'algorithme Smith-Waterman utilisant la technologie SIMD ( single instruction, multiple data ) disponible dans les processeurs Intel Pentium MMX et des technologies similaires a été décrite dans une publication de Rognes et Seeberg. Contrairement à l'approche de Wozniak (1997), la nouvelle implémentation était basée sur des vecteurs parallèles à la séquence de requête, et non sur des vecteurs diagonaux. La société Sencel Bioinformatics a déposé une demande de brevet couvrant cette approche. Sencel continue de développer le logiciel et fournit gratuitement des exécutables pour une utilisation académique.
Une vectorisation SSE2 de l'algorithme (Farrar, 2007) est désormais disponible, offrant une accélération de 8 à 16 fois sur les processeurs Intel/AMD avec extensions SSE2. Lorsqu'elle est exécutée sur un processeur Intel utilisant la microarchitecture Core, l'implémentation SSE2 atteint une augmentation de 20 fois. L'implémentation SSE2 de Farrar est disponible sous forme de programme SSEARCH dans le package de comparaison de séquences FASTA . Le programme SSEARCH est inclus dans la suite de programmes de recherche de similarité de l'Institut européen de bioinformatique .
La société danoise de bioinformatique CLC bio a obtenu des accélérations de près de 200 par rapport aux implémentations logicielles standard avec SSE2 sur un processeur Intel Core 2 Duo à 2,17 GHz, selon un livre blanc accessible au public.
La version accélérée de l'algorithme Smith-Waterman, sur les serveurs Linux basés sur Intel et Advanced Micro Devices (AMD) , est prise en charge par le package GenCore 6, proposé par Biocceleration. Les tests de performances de ce package logiciel montrent une accélération de la vitesse jusqu'à 10 fois supérieure à celle de l'implémentation logicielle standard sur le même processeur.
Actuellement seule entreprise en bioinformatique à proposer à la fois des solutions SSE et FPGA accélérant Smith-Waterman, CLC bio a obtenu des accélérations de plus de 110 par rapport aux implémentations logicielles standard avec CLC Bioinformatics Cube.
L'implémentation la plus rapide de l'algorithme sur les processeurs avec SSSE3 se trouve dans le logiciel SWIPE (Rognes, 2011), qui est disponible sous la licence publique générale GNU Affero . En parallèle, ce logiciel compare les résidus de seize séquences de bases de données différentes à un résidu de requête. En utilisant une séquence de requête de 375 résidus, une vitesse de 106 milliards de mises à jour de cellules par seconde (GCUPS) a été atteinte sur un système à double processeur Intel Xeon X5650 à six cœurs, ce qui est plus de six fois plus rapide que le logiciel basé sur l'approche « striped » de Farrar. Il est plus rapide que BLAST lorsqu'il utilise la matrice BLOSUM50.
Une implémentation de Smith–Waterman appelée diagonalsw , en C et C++ , utilise les jeux d'instructions SIMD ( SSE4.1 pour la plateforme x86 et AltiVec pour la plateforme PowerPC). Elle est publiée sous une licence MIT open source .
Moteur de téléphonie mobile à large bande
En 2008, Farrar a décrit un portage du Striped Smith–Waterman sur le Cell Broadband Engine et a signalé des vitesses de 32 et 12 GCUPS sur une lame IBM QS20 et une Sony PlayStation 3 , respectivement.
Limites
L'expansion rapide des données génétiques met à rude épreuve la rapidité des algorithmes actuels d'alignement des séquences d'ADN. Les besoins essentiels en matière de méthode efficace et précise de découverte de variantes d'ADN exigent des approches innovantes pour un traitement parallèle en temps réel.