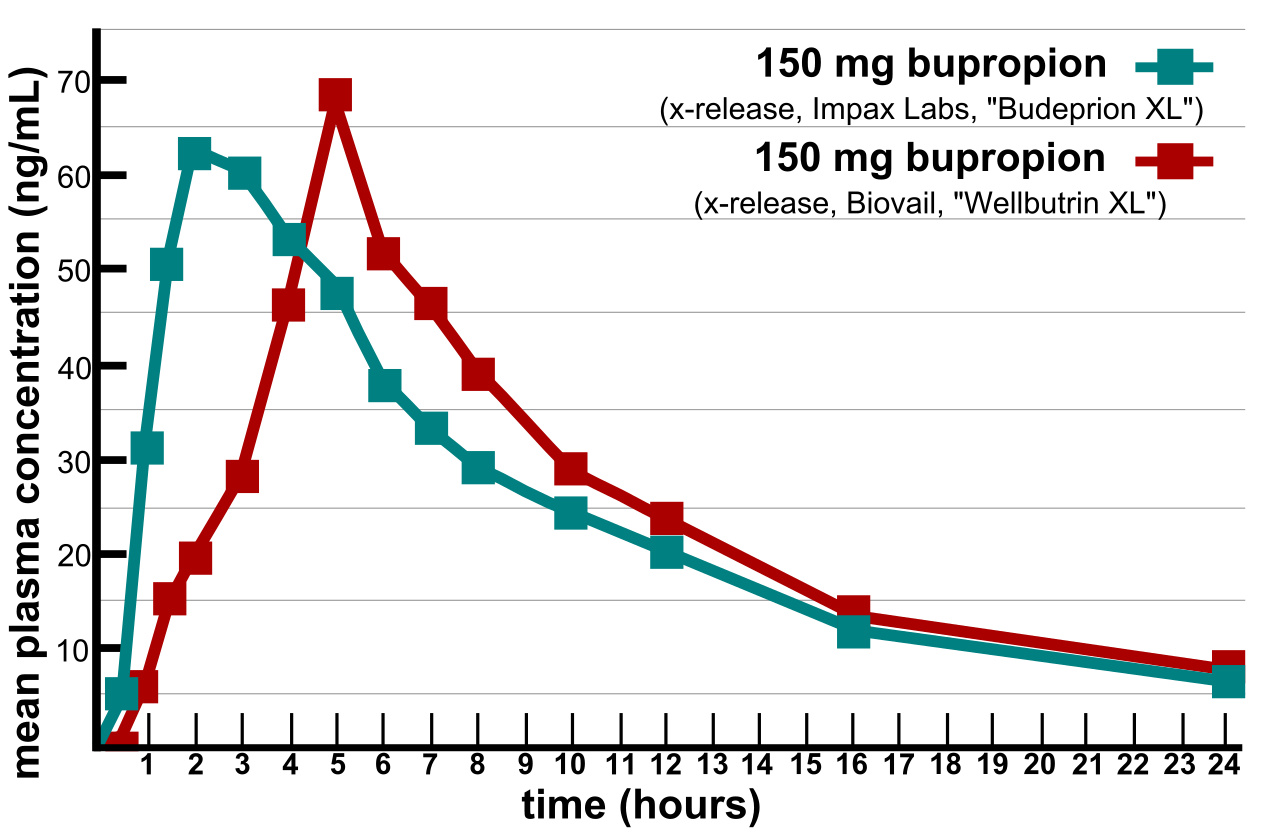
La bioéquivalence est un terme utilisé en pharmacocinétique pour évaluer l' équivalence biologique in vivo attendue de deux préparations brevetées d'un médicament. Si deux produits sont dits bioéquivalents, cela signifie qu'ils devraient être, à toutes fins utiles, identiques.
Un article définit la bioéquivalence en affirmant que « deux produits pharmaceutiques sont bioéquivalents s'ils sont pharmaceutiquement équivalents et que leurs biodisponibilités (taux et degré de disponibilité) après administration à la même dose molaire sont similaires à un degré tel que leurs effets, en termes d' efficacité et de sécurité, peuvent être considérés comme essentiellement les mêmes. L'équivalence pharmaceutique implique la même quantité de la ou des mêmes substances actives, dans la même forme posologique, pour la même voie d'administration et répondant à des normes identiques ou comparables. »
Pour l'Organisation mondiale de la santé (OMS), « deux produits pharmaceutiques sont bioéquivalents s'ils sont pharmaceutiquement équivalents ou des alternatives pharmaceutiques, et que leurs biodisponibilités, en termes de taux (Cmax et tmax) et de degré d'absorption (aire sous la courbe), après administration de la même dose molaire dans les mêmes conditions, sont similaires à un tel degré que l'on peut s'attendre à ce que leurs effets soient essentiellement les mêmes ».
La Food and Drug Administration (FDA) des États-Unis a défini la bioéquivalence comme « l'absence de différence significative dans la vitesse et la mesure dans laquelle le principe actif ou la fraction active des équivalents pharmaceutiques ou des alternatives pharmaceutiques devient disponible au site d'action du médicament lorsqu'il est administré à la même dose molaire dans des conditions similaires dans une étude conçue de manière appropriée. »
Bioéquivalence
Pour déterminer la bioéquivalence entre deux produits tels qu'un produit de marque disponible dans le commerce et un produit générique potentiellement commercialisé , des études pharmacocinétiques sont menées dans lesquelles chacune des préparations est administrée dans une étude croisée à des sujets volontaires, généralement des individus en bonne santé, mais occasionnellement à des patients. Des échantillons de sérum/plasma sont prélevés à intervalles réguliers et analysés pour déterminer la concentration du médicament parent (ou parfois du métabolite ). Parfois, les niveaux de concentration sanguine ne sont ni réalisables ni possibles pour comparer les deux produits (par exemple, les corticostéroïdes inhalés ), alors les paramètres pharmacodynamiques plutôt que les paramètres pharmacocinétiques (voir ci-dessous) sont utilisés pour la comparaison. Pour une comparaison pharmacocinétique, les données de concentration plasmatique sont utilisées pour évaluer les paramètres pharmacocinétiques clés tels que l'aire sous la courbe (ASC), la concentration maximale ( C max ), le temps jusqu'à la concentration maximale ( t max ) et le temps de latence d'absorption ( t lag ). Les tests doivent être effectués à plusieurs doses différentes, en particulier lorsque le médicament présente une pharmacocinétique non linéaire.
Outre les données issues des études de bioéquivalence, d'autres données peuvent devoir être soumises pour satisfaire aux exigences réglementaires en matière de bioéquivalence. Ces preuves peuvent inclure :
- validation de la méthode analytique
- Études de corrélation in vitro-in vivo ( IVIVC )
Définition réglementaire
L'Organisation mondiale de la santé
L'Organisation mondiale de la santé considère que deux formulations sont bioéquivalentes si l'intervalle de confiance à 90 % pour le rapport produit multisource (générique)/comparateur se situe dans une plage d'acceptation de 80,00 à 125,00 % pour l'ASC 0–t et la C max . Pour les produits pharmaceutiques finis à forte variabilité, la plage d'acceptation applicable pour la C max peut être étendue (jusqu'à 69,84 à 143,19 %).
Australie
En Australie , la Therapeutics Goods Administration (TGA) considère que les préparations sont bioéquivalentes si les intervalles de confiance à 90 % (IC à 90 %) des rapports de taux, entre les deux préparations, de C max et d'ASC se situent dans la plage de 0,80 à 1,25. Le T max doit également être similaire entre les produits.
Les exigences sont plus strictes pour les médicaments ayant un index thérapeutique étroit et/ou un métabolisme saturable – ainsi, aucun produit générique n’existe sur le marché australien pour la digoxine ou la phénytoïne par exemple.
Europe
Conformément à la réglementation applicable dans l' Espace économique européen deux médicaments sont bioéquivalents s'ils sont pharmaceutiquement équivalents ou alternatifs et si leurs biodisponibilités après administration à la même dose molaire sont similaires à un degré tel que leurs effets, tant en termes d'efficacité que de sécurité, seront essentiellement les mêmes. Cela est considéré comme démontré si les intervalles de confiance à 90 % (IC à 90 %) des ratios de l'ASC 0–t et de la C max entre les deux préparations se situent dans la plage de 80 à 125 %.
États-Unis
La FDA considère que deux produits sont bioéquivalents si l'IC à 90 % de la moyenne relative de la C max , de l'ASC (0–t) et de l'ASC (0–∞) du test (par exemple, formulation générique) par rapport à la référence (par exemple, formulation de marque innovante) doit être compris entre 80 % et 125 % à jeun. Bien qu'il existe quelques exceptions, une comparaison bioéquivalente des formulations du test et de la référence nécessite généralement également l'administration après un repas approprié à une heure spécifiée avant la prise du médicament, une étude dite « d'effet alimentaire » ou « d'effet alimentaire ». Une étude d'effet alimentaire nécessite la même évaluation statistique que l'étude à jeun, décrite ci-dessus.
Chine

En Chine, il n'existait aucune exigence de bioéquivalence pour les médicaments génériques jusqu'à l' avis de 2016 sur la conduite d'une évaluation cohérente de la qualité et de l'efficacité des médicaments génériques (关于开展仿制药质量和疗效一致性评价的意见), qui a établi les règles de base pour les futurs travaux de bioéquivalence. Depuis juillet 2020, tous les génériques nouvellement approuvés doivent passer des contrôles de bioéquivalence ; les médicaments précédents peuvent demander à être contrôlés. Depuis 2019, le système national d'approvisionnement centralisé basé sur le volume utilise « l'évaluation de la cohérence générique » comme l'un des critères d'appel d'offres.
La définition chinoise de la « bioéquivalence » implique que la moyenne géométrique de la C max , de l'ASC (0–t) et de l'ASC (0–∞) du médicament testé se situe entre 80 % et 125 % de celle du médicament de référence à la fois à jeun et après avoir mangé. Le médicament de référence doit être de préférence le médicament de marque d'origine, puis (s'il n'est pas disponible) un générique reconnu internationalement et approuvé par un pays développé, puis (s'il n'est toujours pas disponible) un générique reconnu internationalement et approuvé au niveau national - ceci afin d'éviter tout écart par rapport au médicament d'origine par l'utilisation en série de génériques comme référence. Si les valeurs pharmacocinétiques telles que la C max ne s'appliquent pas au type de médicament (par exemple si le médicament n'est pas absorbé par voie orale), des comparaisons peuvent être effectuées en utilisant d'autres moyens tels que les courbes dose-réponse .
Selon Wei et al. (2022), la politique d'évaluation de la cohérence a augmenté les dépenses de R&D des sociétés pharmaceutiques chinoises, en particulier parmi les sociétés privées et à haut rendement. Liu et al. (2023) soutiennent que la politique a augmenté la qualité de l'innovation de l'industrie pharmaceutique chinoise.
Problèmes de bioéquivalence
Bien que la FDA maintienne que les médicaments génériques approuvés sont équivalents à leurs homologues de marque, des problèmes de bioéquivalence ont été signalés par des médecins et des patients pour de nombreux médicaments. Certaines classes de médicaments sont suspectées d'être particulièrement problématiques en raison de leur composition chimique. Parmi celles-ci figurent les médicaments chiraux , les médicaments mal absorbés et les médicaments cytotoxiques. De plus, des mécanismes d'administration complexes peuvent entraîner des variations de bioéquivalence. Les médecins sont avertis d'éviter de faire passer les patients de médicaments de marque à des médicaments génériques, ou de passer d'un fabricant générique à un autre, lorsqu'ils prescrivent des médicaments antiépileptiques, la warfarine et la lévothyroxine .
Des problèmes majeurs ont été soulevés dans la vérification de la bioéquivalence lorsque plusieurs versions génériques de médicaments génériques approuvés par la FDA se sont avérées ne pas être équivalentes en termes d'efficacité et d'effets secondaires. En 2007, deux fournisseurs d'informations aux consommateurs sur les produits et suppléments nutritionnels, ConsumerLab.com et The People's Pharmacy, ont publié les résultats de tests comparatifs de différentes marques de bupropion. The People's Pharmacy a reçu de nombreux rapports d'effets secondaires accrus et d'efficacité réduite du bupropion générique, ce qui l'a incité à demander à ConsumerLab.com de tester les produits en question. Les tests ont montré que certaines versions génériques de Wellbutrin XL 300 mg n'avaient pas les mêmes performances que la pilule de marque dans les tests de laboratoire. La FDA a enquêté sur ces plaintes et a conclu que la version générique est équivalente à Wellbutrin XL en ce qui concerne la biodisponibilité du bupropion et de son principal métabolite actif, l'hydroxybupropion. La FDA a également déclaré que la variation naturelle de l'humeur par coïncidence est l'explication la plus probable de l'aggravation apparente de la dépression après le passage de Wellbutrin XL à Budeprion XL. Après avoir nié pendant plusieurs années les rapports des patients, en 2012, la FDA a renversé cet avis, annonçant que « Budeprion XL 300 mg ne parvient pas à démontrer l'équivalence thérapeutique avec Wellbutrin XL 300 mg. » La FDA n'a pas testé la bioéquivalence d'aucune des autres versions génériques de Wellbutrin XL 300 mg, mais a demandé aux quatre fabricants de lui soumettre des données sur cette question avant mars 2013. En octobre 2013, la FDA a déterminé que les formulations de certains fabricants n'étaient pas bioéquivalentes.
En 2004, il a été révélé que Ranbaxy falsifiait des données concernant les médicaments génériques qu'elle fabriquait. En conséquence, 30 produits ont été retirés du marché américain et Ranbaxy a dû payer 500 millions de dollars d'amendes. La FDA a enquêté sur de nombreux fabricants de médicaments indiens après cette découverte, et au moins 12 entreprises se sont vu interdire d'expédier des médicaments aux États-Unis.
En 2017, l' Agence européenne des médicaments a recommandé la suspension d'un certain nombre de médicaments approuvés au niveau national pour lesquels des études de bioéquivalence ont été menées par Micro Therapeutic Research Labs en Inde, en raison d'inspections identifiant une fausse représentation des données d'étude et des lacunes dans la documentation et le traitement des données.