Un arbre phylogénétique , la phylogénie ou l'arbre évolutif est une représentation graphique qui montre l' histoire évolutive d'un ensemble d' espèces ou de taxons au cours d'une période donnée. En d'autres termes, il s'agit d'un diagramme de ramification ou d'un arbre montrant les relations évolutives entre diverses espèces biologiques ou d'autres entités en fonction des similitudes et des différences dans leurs caractéristiques physiques ou génétiques. En biologie évolutive, toute vie sur Terre fait théoriquement partie d'un seul arbre phylogénétique, indiquant une ascendance commune . La phylogénétique est l'étude des arbres phylogénétiques. Le principal défi est de trouver un arbre phylogénétique représentant une ascendance évolutive optimale entre un ensemble d'espèces ou de taxons. La phylogénétique computationnelle (également l'inférence phylogénétique) se concentre sur les algorithmes impliqués dans la recherche d'un arbre phylogénétique optimal dans le paysage phylogénétique.
Les arbres phylogénétiques peuvent être enracinés ou non. Dans un arbre phylogénétique enraciné , chaque nœud avec des descendants représente l' ancêtre commun le plus récent déduit de ces descendants, et les longueurs des arêtes dans certains arbres peuvent être interprétées comme des estimations de temps. Chaque nœud est appelé unité taxonomique. Les nœuds internes sont généralement appelés unités taxonomiques hypothétiques, car ils ne peuvent pas être observés directement. Les arbres sont utiles dans les domaines de la biologie tels que la bioinformatique , la systématique et la phylogénétique . Les arbres non enracinés illustrent uniquement la parenté des nœuds foliaires et ne nécessitent pas que la racine ancestrale soit connue ou déduite.
Histoire
L'idée d'un arbre de vie est née de notions anciennes de progression en échelle depuis les formes de vie inférieures vers les formes de vie supérieures (comme dans la Grande Chaîne de l'Être ). Les premières représentations d'arbres phylogénétiques « ramifiés » comprennent un « tableau paléontologique » montrant les relations géologiques entre les plantes et les animaux dans le livre Géologie élémentaire , d' Edward Hitchcock (première édition : 1840).
Charles Darwin a présenté un « arbre » évolutif schématique dans son livre De l'origine des espèces paru en 1859. Plus d'un siècle plus tard, les biologistes évolutionnistes utilisent encore des diagrammes arborescents pour décrire l'évolution , car ces diagrammes transmettent efficacement le concept selon lequel la spéciation se produit par le biais d'une division adaptative et semi-aléatoire des lignées.
Le terme phylogénétique , ou phylogénie , dérive des deux mots grecs anciens φῦλον ( phûlon ), qui signifie « race, lignée », et γένεσις ( génesis ), qui signifie « origine, source ».
Propriétés
Arbre enraciné
Un arbre phylogénétique enraciné (voir les deux graphiques ci-dessus) est un arbre orienté avec un nœud unique — la racine — correspondant à l'ancêtre commun le plus récent (généralement imputé ) de toutes les entités situées aux feuilles de l'arbre. Le nœud racine n'a pas de nœud parent, mais sert de parent à tous les autres nœuds de l'arbre. La racine est donc un nœud de degré 2, tandis que les autres nœuds internes ont un degré minimum de 3 (où « degré » fait ici référence au nombre total d'arêtes entrantes et sortantes).
La méthode la plus courante pour enraciner les arbres est l'utilisation d'un groupe externe non controversé, suffisamment proche pour permettre une inférence à partir de données de caractéristiques ou d'un séquençage moléculaire, mais suffisamment éloigné pour constituer un groupe externe clair. Une autre méthode est l'enracinement au point médian, ou un arbre peut également être enraciné en utilisant un modèle de substitution non stationnaire .
Arbre déraciné
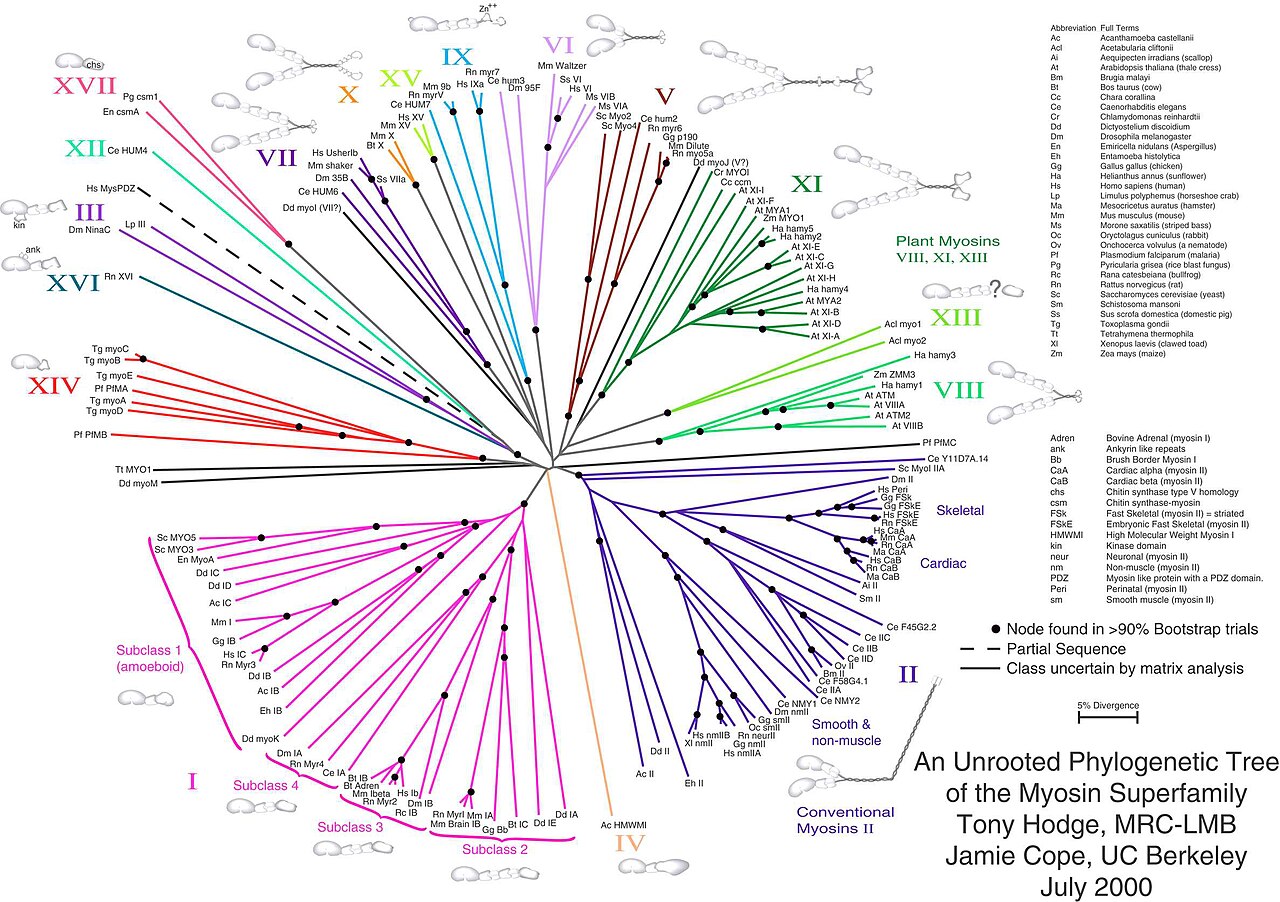
Les arbres sans racines illustrent la parenté des nœuds de feuille sans faire d'hypothèses sur l'ascendance. Ils ne nécessitent pas que la racine ancestrale soit connue ou déduite. Les arbres sans racines peuvent toujours être générés à partir d'arbres enracinés en omettant simplement la racine. En revanche, déduire la racine d'un arbre sans racines nécessite un moyen d'identifier l'ascendance. Cela se fait normalement en incluant un groupe externe dans les données d'entrée de sorte que la racine se trouve nécessairement entre le groupe externe et le reste des taxons de l'arbre, ou en introduisant des hypothèses supplémentaires sur les taux relatifs d'évolution sur chaque branche, comme une application de l' hypothèse de l' horloge moléculaire .
Bifurcation versus multifurcation
Les arbres enracinés et non enracinés peuvent être bifurqués ou multifurqués. Un arbre bifurqué enraciné a exactement deux descendants issus de chaque nœud intérieur (c'est-à-dire qu'il forme un arbre binaire ), et un arbre bifurqué non enraciné prend la forme d'un arbre binaire non enraciné , un arbre libre avec exactement trois voisins à chaque nœud interne. En revanche, un arbre multifurqué enraciné peut avoir plus de deux enfants à certains nœuds et un arbre multifurqué non enraciné peut avoir plus de trois voisins à certains nœuds.
Étiqueté versus non étiqueté
Les arbres enracinés et non enracinés peuvent être étiquetés ou non étiquetés. Un arbre étiqueté a des valeurs spécifiques attribuées à ses feuilles, tandis qu'un arbre non étiqueté, parfois appelé forme d'arbre, définit uniquement une topologie. Certains arbres basés sur des séquences construits à partir d'un petit locus génomique, comme Phylotree, présentent des nœuds internes étiquetés avec des haplotypes ancestraux déduits.
Dénombrer les arbres

Le nombre d'arbres possibles pour un nombre donné de nœuds feuilles dépend du type spécifique d'arbre, mais il y a toujours plus d'arbres étiquetés que d'arbres non étiquetés, plus d'arbres multifurcants que d'arbres bifurcants et plus d'arbres enracinés que d'arbres non enracinés. La dernière distinction est la plus pertinente biologiquement ; elle survient parce qu'il existe de nombreux endroits sur un arbre non enraciné où placer la racine. Pour les arbres bifurcants étiquetés, le nombre total d'arbres enracinés est :
Pour les arbres étiquetés bifurqués, le nombre total d'arbres non enracinés est :
Parmi les arbres bifurquants étiquetés, le nombre d'arbres non enracinés avec des feuilles est égal au nombre d'arbres enracinés avec des feuilles.
Le nombre d'arbres enracinés augmente rapidement en fonction du nombre de pointes. Pour 10 pointes, il y a plus d'arbres bifurquants que possible, et le nombre d'arbres multifurquants augmente plus rapidement, avec environ 7 fois plus de ces derniers que de ceux des premiers.
Types d'arbres spéciaux
Dendrogramme
Un dendrogramme est un nom général pour un arbre, qu'il soit phylogénétique ou non, et donc également pour la représentation schématique d'un arbre phylogénétique.
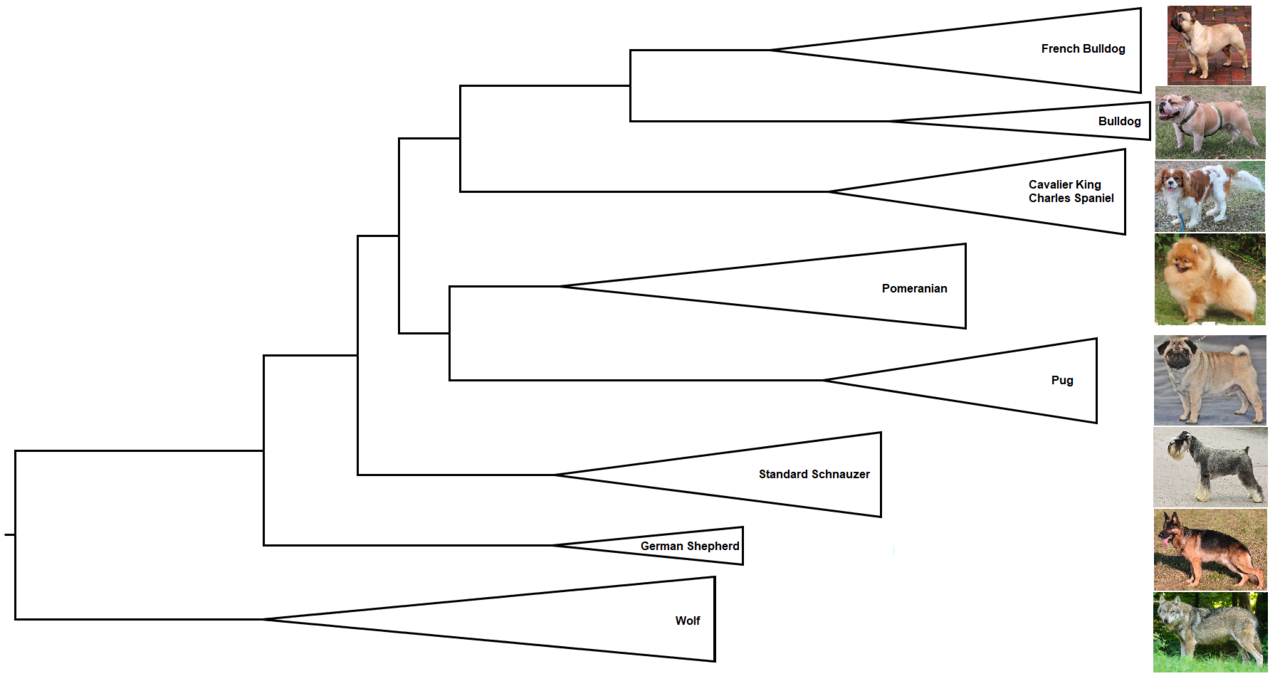
Cladogramme
Un cladogramme ne représente qu'un modèle de ramification ; c'est-à-dire que la longueur de ses branches ne représente pas le temps ou la quantité relative de changement de caractère, et ses nœuds internes ne représentent pas les ancêtres.
Phylogramme
Un phylogramme est un arbre phylogénétique dont les longueurs de branches sont proportionnelles à la quantité de changement de caractère.
Chronogramme
Un chronogramme est un arbre phylogénétique qui représente explicitement le temps à travers la longueur de ses branches.

Dahlgrenogramme
Un dahlgrenogramme est un diagramme représentant une coupe transversale d'un arbre phylogénétique.
Réseau phylogénétique
Un réseau phylogénétique n'est pas à proprement parler un arbre, mais plutôt un graphe plus général , ou un graphe acyclique orienté dans le cas des réseaux enracinés. Ils permettent de surmonter certaines limitations inhérentes aux arbres.
Diagramme de broche
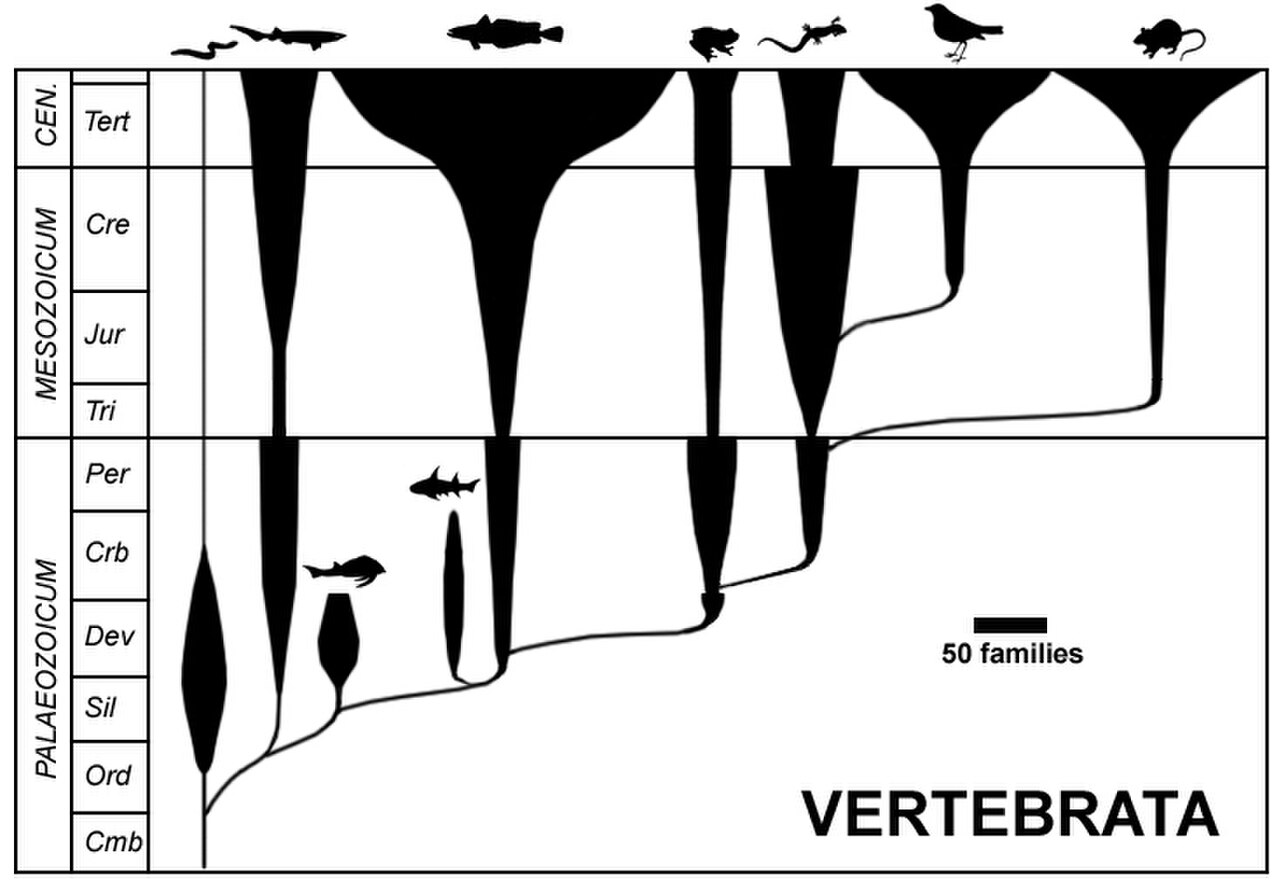
Un diagramme en fuseau, ou diagramme à bulles, est souvent appelé romérogramme, après sa popularisation par le paléontologue américain Alfred Romer . Il représente la diversité taxonomique (largeur horizontale) par rapport au temps géologique (axe vertical) afin de refléter la variation de l'abondance de divers taxons au fil du temps. Un diagramme en fuseau n'est pas un arbre évolutif : les fuseaux taxonomiques obscurcissent les relations réelles du taxon parent au taxon fille et ont l'inconvénient d'impliquer la paraphylie du groupe parental. Ce type de diagramme n'est plus utilisé sous la forme initialement proposée.
Corail de la vie

Darwin a également mentionné que le corail pourrait être une métaphore plus appropriée que l' arbre . En effet, les coraux phylogénétiques sont utiles pour décrire la vie passée et présente, et ils présentent certains avantages par rapport aux arbres ( anastomoses autorisées, etc.).
Construction
Les arbres phylogénétiques composés d'un nombre non négligeable de séquences d'entrée sont construits à l'aide de méthodes de phylogénétique computationnelle . Les méthodes de matrice de distance telles que neighbor-joining ou UPGMA , qui calculent la distance génétique à partir de plusieurs alignements de séquences , sont les plus simples à mettre en œuvre, mais n'invoquent pas de modèle évolutif. De nombreuses méthodes d'alignement de séquences telles que ClustalW créent également des arbres en utilisant les algorithmes les plus simples (c'est-à-dire ceux basés sur la distance) de construction d'arbres. La parcimonie maximale est une autre méthode simple d'estimation des arbres phylogénétiques, mais implique un modèle implicite d'évolution (c'est-à-dire la parcimonie). Des méthodes plus avancées utilisent le critère d'optimalité du maximum de vraisemblance , souvent dans un cadre bayésien , et appliquent un modèle explicite d'évolution à l'estimation des arbres phylogénétiques. L'identification de l'arbre optimal à l'aide de plusieurs de ces techniques est NP-difficile , donc des méthodes de recherche heuristique et d'optimisation sont utilisées en combinaison avec des fonctions de notation d'arbre pour identifier un arbre raisonnablement bon qui correspond aux données.
Les méthodes de construction d’arbres peuvent être évaluées sur la base de plusieurs critères :
- efficacité (combien de temps faut-il pour calculer la réponse, combien de mémoire faut-il ?)
- puissance (fait-on bon usage des données ou les informations sont-elles gaspillées ?)
- cohérence (convergera-t-elle vers la même réponse à plusieurs reprises, si à chaque fois des données différentes sont données pour le même problème de modèle ?)
- robustesse (résiste-t-il bien aux violations des hypothèses du modèle sous-jacent ?)
- falsifiabilité (nous alerte-t-elle lorsqu'il n'est pas bon de l'utiliser, c'est-à-dire lorsque des hypothèses sont violées ?)
Les techniques de construction d'arbres ont également retenu l'attention des mathématiciens. Les arbres peuvent également être construits en utilisant la théorie T.
Formats de fichiers
Les arbres peuvent être codés dans un certain nombre de formats différents, qui doivent tous représenter la structure imbriquée d'un arbre. Ils peuvent ou non coder les longueurs de branches et d'autres caractéristiques. Les formats standardisés sont essentiels pour distribuer et partager des arbres sans avoir recours à une sortie graphique difficile à importer dans un logiciel existant. Les formats couramment utilisés sont
Limites de l’analyse phylogénétique
Bien que les arbres phylogénétiques produits à partir de gènes séquencés ou de données génomiques de différentes espèces puissent fournir des informations sur l'évolution, ces analyses présentent d'importantes limites. Plus important encore, les arbres qu'ils génèrent ne sont pas nécessairement corrects : ils ne représentent pas nécessairement avec précision l'histoire évolutive des taxons inclus. Comme tout résultat scientifique, ils sont sujets à falsification par des études plus poussées (par exemple, la collecte de données supplémentaires, l'analyse des données existantes avec des méthodes améliorées). Les données sur lesquelles ils sont basés peuvent être bruitées ; l'analyse peut être faussée par la recombinaison génétique , le transfert horizontal de gènes , l'hybridation entre des espèces qui n'étaient pas les plus proches voisines sur l'arbre avant l'hybridation et les séquences conservées .
De plus, il est difficile de fonder une analyse sur un seul type de caractère, tel qu'un seul gène ou une seule protéine , ou sur une analyse morphologique uniquement, car de tels arbres construits à partir d'une autre source de données sans rapport avec le premier diffèrent souvent du premier, et il faut donc faire très attention lors de l'inférence des relations phylogénétiques entre les espèces. Cela est particulièrement vrai pour le matériel génétique qui est sujet à un transfert latéral de gènes et à une recombinaison , où différents blocs d'haplotypes peuvent avoir des histoires différentes. Dans ces types d'analyse, l'arbre de sortie d'une analyse phylogénétique d'un seul gène est une estimation de la phylogénie du gène (c'est-à-dire un arbre génétique) et non de la phylogénie des taxons (c'est-à-dire un arbre d'espèces) à partir desquels ces caractères ont été échantillonnés, bien que idéalement, les deux devraient être très proches. Pour cette raison, les études phylogénétiques sérieuses utilisent généralement une combinaison de gènes provenant de différentes sources génomiques (par exemple, des génomes mitochondriaux ou plastidiques par rapport aux génomes nucléaires), ou des gènes qui devraient évoluer sous différents régimes sélectifs, de sorte que l'homoplasie (fausse homologie ) serait peu susceptible de résulter de la sélection naturelle.
Lorsque des espèces éteintes sont incluses comme nœuds terminaux dans une analyse (plutôt que, par exemple, pour contraindre les nœuds internes), elles sont considérées comme ne représentant pas les ancêtres directs d'une espèce existante. Les espèces éteintes ne contiennent généralement pas d'ADN de haute qualité .
La gamme des matériaux ADN utiles s'est élargie grâce aux progrès des technologies d'extraction et de séquençage. Le développement de technologies permettant de déduire des séquences à partir de fragments plus petits ou de modèles spatiaux de produits de dégradation de l'ADN permettrait d'élargir encore la gamme des ADN considérés comme utiles.
Les arbres phylogénétiques peuvent également être déduits d’une gamme d’autres types de données, notamment la morphologie, la présence ou l’absence de types particuliers de gènes, les événements d’insertion et de suppression – et toute autre observation considérée comme contenant un signal évolutif.
Les réseaux phylogénétiques sont utilisés lorsque les arbres bifurquants ne sont pas adaptés, en raison de ces complications qui suggèrent une histoire évolutive plus réticulaire des organismes échantillonnés.