Le criblage virtuel ( VS ) est une technique informatique utilisée dans la découverte de médicaments pour rechercher des bibliothèques de petites molécules afin d'identifier les structures les plus susceptibles de se lier à une cible médicamenteuse , généralement un récepteur protéique ou une enzyme .
Le criblage virtuel a été défini comme « l'évaluation automatique de très grandes bibliothèques de composés » à l'aide de programmes informatiques. Comme le suggère cette définition, le criblage virtuel a été en grande partie un jeu de chiffres se concentrant sur la façon dont l'énorme espace chimique de plus de 10 60 composés concevables peut être filtré en un nombre gérable qui peut être synthétisé, acheté et testé. Bien que la recherche dans l'ensemble de l'univers chimique puisse être un problème théoriquement intéressant, des scénarios de criblage virtuel plus pratiques se concentrent sur la conception et l'optimisation de bibliothèques combinatoires ciblées et l'enrichissement des bibliothèques de composés disponibles à partir de référentiels de composés internes ou d'offres de fournisseurs. À mesure que la précision de la méthode a augmenté, le criblage virtuel est devenu partie intégrante du processus de découverte de médicaments . Le criblage virtuel peut être utilisé pour sélectionner des composés de bases de données internes à cribler, choisir des composés qui peuvent être achetés en externe et choisir quel composé doit être synthétisé ensuite.
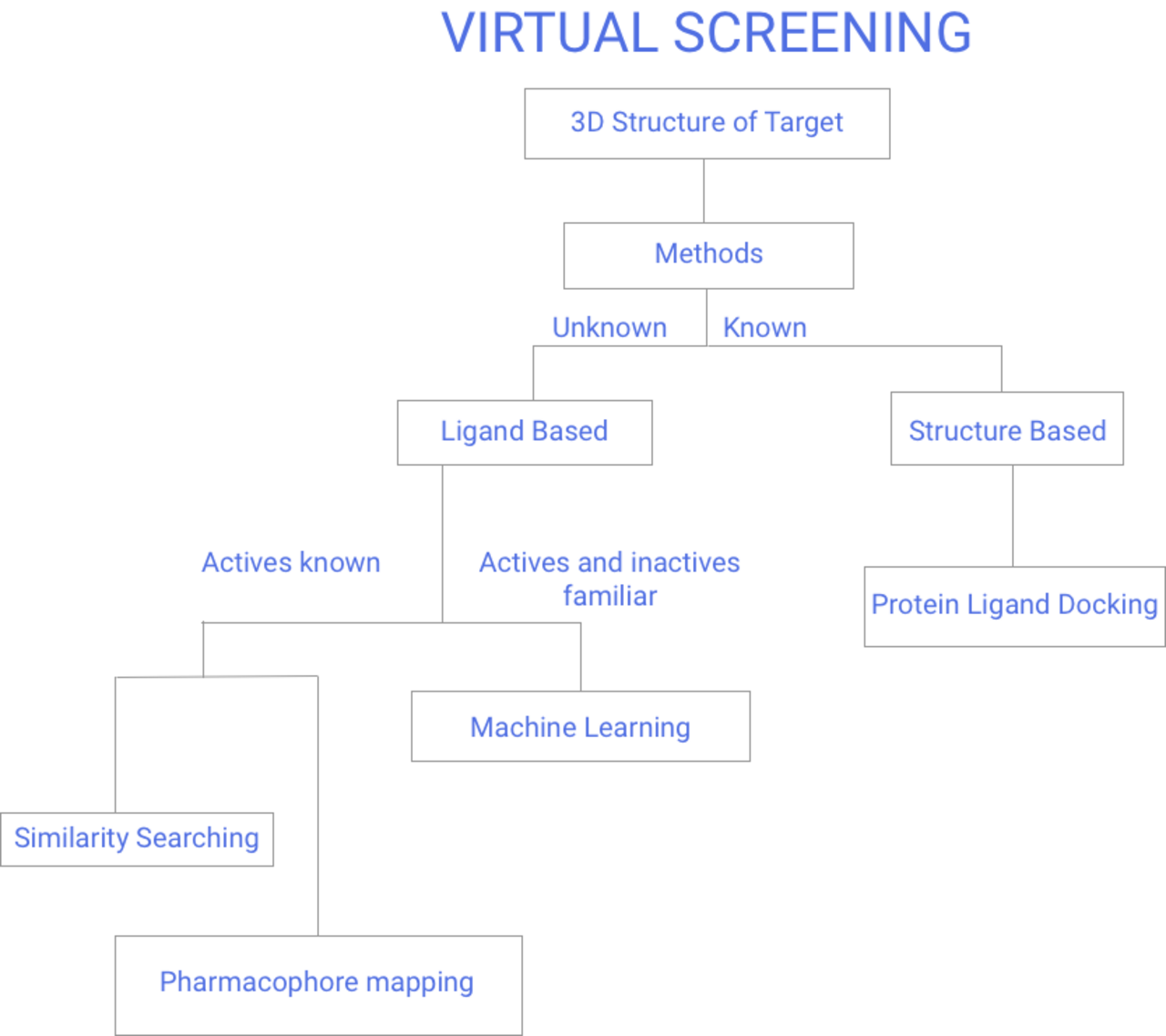
Méthodes
Il existe deux grandes catégories de techniques de criblage : basées sur les ligands et basées sur la structure. Le reste de cette page reflétera le diagramme de flux de la figure 1 du criblage virtuel.
Méthodes basées sur les ligands
Étant donné un ensemble de ligands structurellement divers qui se lient à un récepteur , un modèle du récepteur peut être construit en exploitant les informations collectives contenues dans cet ensemble de ligands. Différentes techniques de calcul explorent les similitudes structurelles, électroniques, de forme moléculaire et physicochimiques de différents ligands qui pourraient impliquer leur mode d'action contre un récepteur moléculaire spécifique ou des lignées cellulaires. Un ligand candidat peut ensuite être comparé au modèle pharmacophore pour déterminer s'il est compatible avec celui-ci et donc susceptible de se lier. Différentes méthodes d'analyse de similarité chimique 2D ont été utilisées pour analyser une base de données afin de trouver des ligands actifs. Une autre approche populaire utilisée dans le criblage virtuel basé sur les ligands consiste à rechercher des molécules de forme similaire à celle des actifs connus, car ces molécules s'adapteront au site de liaison de la cible et seront donc susceptibles de se lier à la cible. Il existe un certain nombre d'applications prospectives de cette classe de techniques dans la littérature. Des extensions pharmacophores de ces méthodes 3D sont également disponibles gratuitement sous forme de serveurs Web. Le criblage virtuel basé sur la forme a également gagné en popularité.
Méthodes basées sur la structure
L'approche de criblage virtuel basée sur la structure comprend différentes techniques informatiques qui prennent en compte la structure du récepteur qui est la cible moléculaire des ligands actifs étudiés. Certaines de ces techniques comprennent l'amarrage moléculaire , la prédiction pharmacophore basée sur la structure et les simulations de dynamique moléculaire. L'amarrage moléculaire est la technique basée sur la structure la plus utilisée, et il applique une fonction de notation pour estimer l'aptitude de chaque ligand par rapport au site de liaison du récepteur macromoléculaire, aidant à choisir les ligands ayant la plus grande affinité. Actuellement, il existe des serveurs Web orientés vers le criblage virtuel prospectif.
Méthodes hybrides
Des méthodes hybrides qui s'appuient sur la similarité structurelle et ligand ont également été développées pour surmonter les limites des approches VLS traditionnelles. Cette méthodologie utilise des informations de liaison ligand-ligand basées sur l'évolution pour prédire les liants de petites molécules et peut utiliser à la fois la similarité structurelle globale et la similarité de poche. Une approche basée sur la similarité structurelle globale utilise à la fois une structure expérimentale ou un modèle protéique prédit pour trouver une similarité structurelle avec des protéines dans la bibliothèque de modèles holo-PDB. Lors de la détection d'une similarité structurelle significative, une métrique de coefficient Tanimoto basée sur l'empreinte digitale 2D est appliquée pour rechercher de petites molécules similaires aux ligands extraits de modèles holo-PDB sélectionnés. Les prédictions de cette méthode ont été évaluées expérimentalement et montrent un bon enrichissement dans l'identification de petites molécules actives.
La méthode spécifiée ci-dessus dépend de la similarité structurelle globale et n'est pas capable de sélectionner a priori un site de liaison ligand-protéine particulier dans la protéine d'intérêt. De plus, comme les méthodes reposent sur une évaluation de la similarité 2D pour les ligands, elles ne sont pas capables de reconnaître la similarité stéréochimique de petites molécules qui sont sensiblement différentes mais présentent une similarité de forme géométrique. Pour répondre à ces préoccupations, une nouvelle approche centrée sur les poches, PoLi, capable de cibler des poches de liaison spécifiques dans des modèles d'holoprotéines, a été développée et évaluée expérimentalement.
Infrastructure informatique
Le calcul des interactions par paires entre atomes, qui est une condition préalable au fonctionnement de nombreux programmes de criblage virtuel, évolue selon , N étant le nombre d'atomes dans le système. En raison de l'échelle quadratique, les coûts de calcul augmentent rapidement.
Approche basée sur les ligands
Les méthodes basées sur les ligands nécessitent généralement une fraction de seconde pour une seule opération de comparaison de structure. Parfois, un seul processeur suffit pour effectuer un criblage de grande envergure en quelques heures. Cependant, plusieurs comparaisons peuvent être effectuées en parallèle afin d'accélérer le traitement d'une grande base de données de composés.
Approche basée sur la structure
La taille de la tâche nécessite une infrastructure informatique parallèle , telle qu'un cluster de systèmes Linux , exécutant un processeur de file d'attente par lots pour gérer le travail, tel que Sun Grid Engine ou Torque PBS.
Il est nécessaire de disposer d'un moyen de gérer les entrées provenant de grandes bibliothèques de composés. Cela nécessite une forme de base de données de composés qui peut être interrogée par le cluster parallèle, fournissant des composés en parallèle aux différents nœuds de calcul. Les moteurs de base de données commerciaux peuvent être trop lourds, et un moteur d'indexation à grande vitesse, tel que Berkeley DB , peut être un meilleur choix. De plus, il peut ne pas être efficace d'exécuter une comparaison par tâche, car le temps de montée en puissance des nœuds du cluster pourrait facilement dépasser la quantité de travail utile. Pour contourner ce problème, il est nécessaire de traiter des lots de composés dans chaque tâche du cluster, en agrégeant les résultats dans une sorte de fichier journal. Un processus secondaire, pour exploiter les fichiers journaux et extraire les candidats à score élevé, peut ensuite être exécuté une fois l'expérience terminée.
Précision
L'objectif du criblage virtuel est d'identifier des molécules de nouvelle structure chimique qui se lient à la cible macromoléculaire d'intérêt . Ainsi, le succès d'un criblage virtuel est défini en termes de découverte de nouveaux échafaudages intéressants plutôt que du nombre total de résultats. Les interprétations de la précision du criblage virtuel doivent donc être considérées avec prudence. De faibles taux de réussite d'échafaudages intéressants sont clairement préférables à des taux de réussite élevés d'échafaudages déjà connus.
La plupart des tests d'études de criblage virtuel dans la littérature sont rétrospectifs. Dans ces études, la performance d'une technique de criblage virtuel est mesurée par sa capacité à récupérer un petit ensemble de molécules déjà connues ayant une affinité avec la cible d'intérêt (molécules actives ou simplement actives) à partir d'une bibliothèque contenant une proportion beaucoup plus élevée de molécules inactives ou de leurres supposés. Il existe plusieurs façons distinctes de sélectionner des leurres en faisant correspondre les propriétés de la molécule active correspondante et plus récemment, les leurres sont également sélectionnés d'une manière sans correspondance de propriétés. L'impact réel de la sélection de leurres, que ce soit à des fins de formation ou de test, a également été discuté.
En revanche, dans les applications prospectives du criblage virtuel, les résultats obtenus sont soumis à une confirmation expérimentale (par exemple, mesures de la CI 50 ). Il existe un consensus sur le fait que les repères rétrospectifs ne sont pas de bons prédicteurs des performances prospectives et que, par conséquent, seules les études prospectives constituent une preuve concluante de l'adéquation d'une technique à une cible particulière.
Application à la découverte de médicaments
Le criblage virtuel est une application très utile lorsqu'il s'agit d'identifier des molécules à succès comme point de départ pour la chimie médicinale. À mesure que l'approche de criblage virtuel commence à devenir une technique plus vitale et plus substantielle au sein de l'industrie de la chimie médicinale, cette approche a connu une croissance rapide.
Méthodes basées sur les ligands
Sans connaître la structure, on essaie de prédire comment les ligands se lieront au récepteur. Grâce à l'utilisation de caractéristiques pharmacophores, chaque ligand identifie les donneurs et les accepteurs. Les caractéristiques d'égalisation sont superposées, mais il est peu probable qu'il existe une seule solution correcte.
Modèles de pharmacophores
Cette technique est utilisée lors de la fusion des résultats de recherches en utilisant des composés de référence différents, des descripteurs et des coefficients identiques, mais des composés actifs différents. Cette technique est avantageuse car elle est plus efficace que l'utilisation d'une seule structure de référence et offre les performances les plus précises lorsqu'il s'agit de composés actifs divers.
Un pharmacophore est un ensemble de caractéristiques stériques et électroniques nécessaires pour avoir une ou des interactions supramoléculaires optimales avec une structure cible biologique afin de précipiter sa réponse biologique. Choisissez un représentant comme un ensemble d'actifs, la plupart des méthodes rechercheront des liaisons similaires. Il est préférable d'avoir plusieurs molécules rigides et les ligands doivent être diversifiés, en d'autres termes, assurez-vous d'avoir des caractéristiques différentes qui n'apparaissent pas pendant la phase de liaison.
Projection virtuelle basée sur la forme
Les approches de similarité moléculaire basées sur la forme sont devenues des techniques de criblage virtuel importantes et populaires. À l'heure actuelle, la plate-forme de criblage hautement optimisée ROCS (Rapid Overlay of Chemical Structures) est considérée comme la norme industrielle de facto pour le criblage virtuel centré sur les ligands et basé sur la forme. Elle utilise une fonction gaussienne pour définir les volumes moléculaires de petites molécules organiques. La sélection de la conformation de la requête est moins importante, ce qui rend le criblage basé sur la forme idéal pour la modélisation basée sur les ligands : la disponibilité d'une conformation bioactive pour la requête n'est pas le facteur limitant du criblage, c'est plutôt la sélection du ou des composés de la requête qui est décisive pour les performances du criblage. D'autres méthodes de similarité moléculaire basées sur la forme telles que Blaze (anciennement connu sous le nom de FieldScreen) et Autodock-SS ont également été développées.
Dépistage virtuel sur le terrain
En tant qu'amélioration des méthodes de similarité basées sur la forme, les méthodes basées sur le champ tentent de prendre en compte tous les champs qui influencent une interaction ligand-récepteur tout en étant indépendantes de la structure chimique utilisée comme requête. Les champs électrostatiques ou hydrophobes sont des exemples d'autres champs utilisés dans ces méthodes.
Relation quantitative-structure-activité
Les modèles de relation quantitative structure-activité (QSAR) sont des modèles prédictifs basés sur des informations extraites d'un ensemble de composés actifs et inactifs connus. Les SAR (Structure Activity Relationship) où les données sont traitées qualitativement et peuvent être utilisées avec des classes structurelles et plus d'un mode de liaison. Les modèles donnent la priorité aux composés pour la découverte de pistes.
Algorithmes d'apprentissage automatique
Les algorithmes d'apprentissage automatique ont été largement utilisés dans les approches de criblage virtuel. Les techniques d'apprentissage supervisé utilisent des ensembles de données d'entraînement et de test composés de composés connus actifs et inactifs. Différents algorithmes d'apprentissage automatique ont été appliqués avec succès dans les stratégies de criblage virtuel, telles que le partitionnement récursif, les machines à vecteurs de support , la forêt aléatoire, les k-plus proches voisins et les réseaux neuronaux . Ces modèles trouvent la probabilité qu'un composé soit actif, puis classent chaque composé en fonction de sa probabilité.
Analyse sous-structurelle en apprentissage automatique
Le premier modèle d'apprentissage automatique utilisé sur de grands ensembles de données est l'analyse de sous-structure qui a été créée en 1973. Chaque fragment de sous-structure apporte une contribution continue à une activité de type spécifique. La sous-structure est une méthode qui surmonte la difficulté de la dimensionnalité massive lorsqu'il s'agit d'analyser des structures dans la conception de médicaments. Une analyse de sous-structure efficace est utilisée pour les structures qui présentent des similitudes avec un bâtiment à plusieurs niveaux ou une tour. La géométrie est utilisée pour numéroter les joints limites d'une structure donnée au début et vers le point culminant. Lorsque la méthode des routines spéciales de condensation statique et de substitution est développée, cette méthode s'avère plus productive que les modèles d'analyse de sous-structure précédents.
Partitionnement récursif
Le partitionnement récursif est une méthode qui crée un arbre de décision à l'aide de données qualitatives. Comprendre la façon dont les règles décomposent les classes avec une faible erreur de classification erronée tout en répétant chaque étape jusqu'à ce qu'aucune division raisonnable ne puisse être trouvée. Cependant, le partitionnement récursif peut avoir une faible capacité de prédiction et créer potentiellement des modèles fins au même rythme.
Méthodes basées sur la structure connues pour l'amarrage des ligands protéiques
Le ligand peut se lier à un site actif au sein d'une protéine en utilisant un algorithme de recherche d'amarrage et une fonction de notation afin d'identifier la cause la plus probable pour un ligand individuel tout en attribuant un ordre de priorité.