En physique et chimie computationnelles , la méthode Hartree-Fock ( HF ) est une méthode d'approximation pour la détermination de la fonction d'onde et de l'énergie d'un système quantique à plusieurs corps dans un état stationnaire .
La méthode Hartree-Fock suppose souvent que la fonction d'onde exacte à N corps du système peut être approximée par un seul déterminant de Slater (dans le cas où les particules sont des fermions ) ou par un seul permanent (dans le cas des bosons ) de N orbitales de spin . En invoquant la méthode variationnelle , on peut dériver un ensemble d' équations couplées à N pour les N orbitales de spin. Une solution de ces équations donne la fonction d'onde Hartree-Fock et l'énergie du système. L'approximation Hartree-Fock est un exemple de théorie du champ moyen , où le fait de négliger les fluctuations d'ordre supérieur dans le paramètre d'ordre permet de remplacer les termes d'interaction par des termes quadratiques, obtenant ainsi des hamiltoniens exactement solubles.
En particulier dans la littérature ancienne, la méthode Hartree-Fock est également appelée méthode du champ auto-cohérent ( SCF ). En dérivant ce qu'on appelle aujourd'hui l' équation de Hartree comme solution approximative de l' équation de Schrödinger , Hartree exigeait que le champ final calculé à partir de la distribution de charge soit « auto-cohérent » avec le champ initial supposé. Ainsi, l'auto-cohérence était une exigence de la solution. Les solutions aux équations non linéaires de Hartree-Fock se comportent également comme si chaque particule était soumise au champ moyen créé par toutes les autres particules (voir l' opérateur de Fock ci-dessous), et la terminologie a donc perduré. Les équations sont presque universellement résolues au moyen d'une méthode itérative , bien que l' algorithme d'itération à virgule fixe ne converge pas toujours. Ce schéma de solution n'est pas le seul possible et n'est pas une caractéristique essentielle de la méthode Hartree-Fock.
La méthode Hartree-Fock trouve son application typique dans la résolution de l'équation de Schrödinger pour les atomes, les molécules, les nanostructures et les solides, mais elle a également trouvé une utilisation répandue en physique nucléaire . (Voir la méthode Hartree-Fock-Bogoliubov pour une discussion de son application en théorie de la structure nucléaire ). Dans la théorie de la structure atomique , les calculs peuvent être effectués pour un spectre avec de nombreux niveaux d'énergie excités, et par conséquent, la méthode Hartree-Fock pour les atomes suppose que la fonction d'onde est une fonction d'état de configuration unique avec des nombres quantiques bien définis et que le niveau d'énergie n'est pas nécessairement l' état fondamental .
Pour les atomes comme pour les molécules, la solution Hartree-Fock constitue le point de départ central de la plupart des méthodes qui décrivent plus précisément le système à plusieurs électrons.
Le reste de cet article se concentrera sur les applications en théorie de la structure électronique adaptées aux molécules, l'atome étant un cas particulier. La discussion ici ne concerne que la méthode Hartree-Fock restreinte, où l'atome ou la molécule est un système à couche fermée avec toutes les orbitales (atomiques ou moléculaires) doublement occupées. Les systèmes à couche ouverte , où certains des électrons ne sont pas appariés, peuvent être traités soit par la méthode Hartree-Fock restreinte à couche ouverte , soit par la méthode Hartree-Fock non restreinte .
Bref historique
Les premières méthodes semi-empiriques
L'origine de la méthode Hartree-Fock remonte à la fin des années 1920, peu après la découverte de l' équation de Schrödinger en 1926. Les méthodes de Douglas Hartree ont été guidées par certaines méthodes antérieures, semi-empiriques, du début des années 1920 (par E. Fues, RB Lindsay et lui-même) basées sur l' ancienne théorie quantique de Bohr.
Dans le modèle de Bohr de l'atome, l'énergie d'un état de nombre quantique principal n est donnée en unités atomiques par . Il a été observé à partir des spectres atomiques que les niveaux d'énergie des atomes à plusieurs électrons sont bien décrits en appliquant une version modifiée de la formule de Bohr. En introduisant le défaut quantique d comme paramètre empirique, les niveaux d'énergie d'un atome générique ont été bien approximés par la formule , dans le sens où l'on pouvait reproduire assez bien les niveaux de transition observés dans la région des rayons X (par exemple, voir la discussion empirique et la dérivation dans la loi de Moseley ). L'existence d'un défaut quantique non nul a été attribuée à la répulsion électron-électron, qui n'existe clairement pas dans l'atome d'hydrogène isolé. Cette répulsion a entraîné un écrantage partiel de la charge nucléaire nue. Ces premiers chercheurs ont ensuite introduit d'autres potentiels contenant des paramètres empiriques supplémentaires dans l'espoir de mieux reproduire les données expérimentales.
Méthode Hartree
En 1927, DR Hartree a introduit une procédure, qu'il a appelée la méthode du champ auto-cohérent, pour calculer les fonctions d'onde et les énergies approximatives des atomes et des ions. Hartree a cherché à se débarrasser des paramètres empiriques et à résoudre l'équation de Schrödinger à plusieurs corps indépendante du temps à partir de principes physiques fondamentaux, c'est-à -dire ab initio . Sa première méthode de résolution proposée est devenue connue sous le nom de méthode Hartree , ou produit Hartree . Cependant, de nombreux contemporains de Hartree ne comprenaient pas le raisonnement physique derrière la méthode Hartree : elle semblait à beaucoup de gens contenir des éléments empiriques, et son lien avec la solution de l'équation de Schrödinger à plusieurs corps n'était pas clair. Cependant, en 1928, JC Slater et JA Gaunt ont montré indépendamment que la méthode Hartree pouvait être formulée sur une base théorique plus solide en appliquant le principe variationnel à un ansatz (fonction d'onde d'essai) en tant que produit de fonctions à une seule particule.
En 1930, Slater et VA Fock ont indépendamment souligné que la méthode Hartree ne respectait pas le principe d' antisymétrie de la fonction d'onde. La méthode Hartree utilisait le principe d'exclusion de Pauli dans sa formulation la plus ancienne, interdisant la présence de deux électrons dans le même état quantique. Cependant, il s'est avéré que cette méthode était fondamentalement incomplète dans sa négligence des statistiques quantiques .
Hartree-Fock
Une solution au manque d'antisymétrie dans la méthode Hartree est apparue lorsqu'il a été démontré qu'un déterminant de Slater , un déterminant des orbitales à une particule utilisé pour la première fois par Heisenberg et Dirac en 1926, satisfait trivialement la propriété antisymétrique de la solution exacte et constitue donc un ansatz approprié pour l'application du principe variationnel . La méthode Hartree originale peut alors être considérée comme une approximation de la méthode Hartree-Fock en négligeant l'échange . La méthode originale de Fock s'appuyait fortement sur la théorie des groupes et était trop abstraite pour que les physiciens contemporains puissent la comprendre et la mettre en œuvre. En 1935, Hartree a reformulé la méthode pour qu'elle soit plus adaptée aux besoins du calcul.
La méthode Hartree-Fock, malgré son image physiquement plus précise, a été peu utilisée jusqu'à l'avènement des ordinateurs électroniques dans les années 1950 en raison des exigences de calcul beaucoup plus importantes par rapport à la méthode Hartree et aux modèles empiriques. Au départ, la méthode Hartree et la méthode Hartree-Fock étaient toutes deux appliquées exclusivement aux atomes, où la symétrie sphérique du système permettait de simplifier considérablement le problème. Ces méthodes approximatives étaient (et sont) souvent utilisées avec l' approximation du champ central pour imposer la condition que les électrons d'une même couche aient la même partie radiale et pour restreindre la solution variationnelle à une fonction propre de spin . Malgré cela, le calcul manuel d'une solution à l'aide des équations Hartree-Fock pour un atome de taille moyenne était laborieux ; les petites molécules nécessitaient des ressources de calcul bien supérieures à ce qui était disponible avant 1950.
Algorithme de Hartree-Fock
La méthode Hartree-Fock est généralement utilisée pour résoudre l'équation de Schrödinger indépendante du temps pour un atome ou une molécule multiélectronique comme décrit dans l' approximation de Born-Oppenheimer . Comme il n'existe pas de solutions analytiques connues pour les systèmes à plusieurs électrons (il existe des solutions pour les systèmes à un électron tels que les atomes d'hydrogène et le cation hydrogène diatomique ), le problème est résolu numériquement. En raison des non-linéarités introduites par l'approximation Hartree-Fock, les équations sont résolues à l'aide d'une méthode non linéaire telle que l'itération , ce qui donne lieu au nom de « méthode du champ auto-cohérent ».
Approximations
La méthode Hartree-Fock propose cinq simplifications majeures pour traiter cette tâche :
- L' approximation de Born-Oppenheimer est supposée par nature. La fonction d'onde moléculaire complète est en fait une fonction des coordonnées de chacun des noyaux, en plus de celles des électrons.
- En règle générale, les effets relativistes sont complètement négligés. L' opérateur d'impulsion est supposé être complètement non relativiste.
- La solution variationnelle est supposée être une combinaison linéaire d'un nombre fini de fonctions de base , qui sont généralement (mais pas toujours) choisies pour être orthogonales . L'ensemble de base fini est supposé être approximativement complet .
- Chaque fonction propre d'énergie est supposée descriptible par un seul déterminant de Slater , un produit antisymétrisé de fonctions d'onde à un électron (c'est-à-dire d'orbitales ).
- L' approximation du champ moyen est implicite. Les effets découlant des écarts par rapport à cette hypothèse sont négligés. Ces effets sont souvent utilisés collectivement comme définition du terme corrélation électronique . Cependant, l'étiquette « corrélation électronique » au sens strict englobe à la fois la corrélation de Coulomb et la corrélation de Fermi, et cette dernière est un effet de l'échange d'électrons, qui est entièrement pris en compte dans la méthode Hartree-Fock. Énoncée dans cette terminologie, la méthode ne néglige que la corrélation de Coulomb. Cependant, il s'agit d'un défaut important, qui explique (entre autres) l'incapacité de Hartree-Fock à capturer la dispersion de Londres .
La relaxation des deux dernières approximations donne lieu à de nombreuses méthodes dites post-Hartree-Fock .
Optimisation variationnelle des orbitales
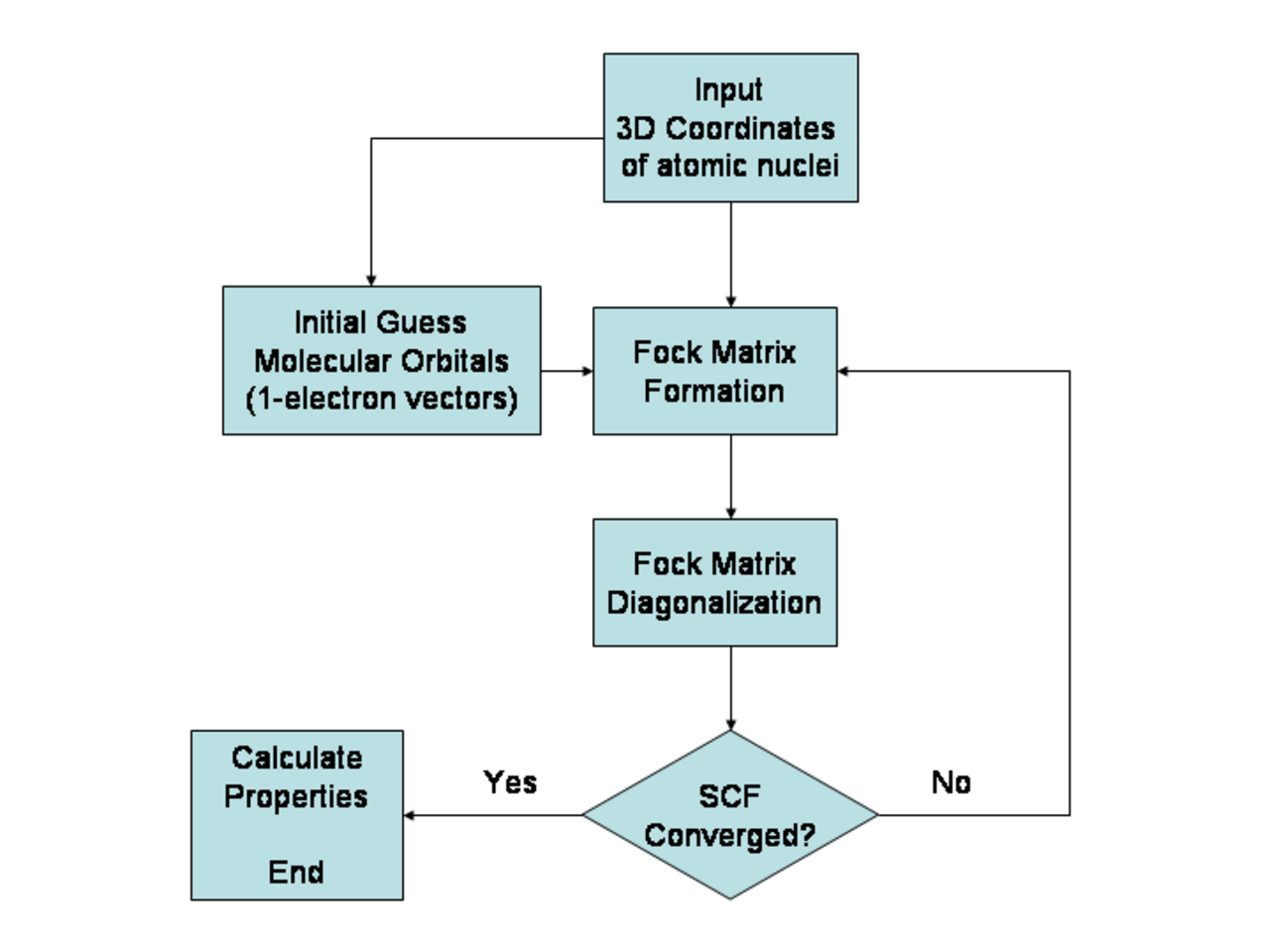
Le théorème variationnel stipule que pour un opérateur hamiltonien indépendant du temps, toute fonction d'onde d'essai aura une valeur d'espérance énergétique supérieure ou égale à la véritable fonction d'onde de l'état fondamental correspondant à l'hamiltonien donné. De ce fait, l'énergie de Hartree-Fock est une limite supérieure de l'énergie de l'état fondamental véritable d'une molécule donnée. Dans le contexte de la méthode Hartree-Fock, la meilleure solution possible se situe à la limite de Hartree-Fock ; c'est-à-dire la limite de l'énergie de Hartree-Fock lorsque l'ensemble de base s'approche de la complétude . (L'autre est la limite de CI complet , où les deux dernières approximations de la théorie de Hartree-Fock telles que décrites ci-dessus sont complètement annulées. Ce n'est que lorsque les deux limites sont atteintes que la solution exacte, jusqu'à l'approximation de Born-Oppenheimer, est obtenue.) L'énergie de Hartree-Fock est l'énergie minimale pour un seul déterminant de Slater.
Le point de départ de la méthode Hartree-Fock est un ensemble de fonctions d'onde à un électron approximatives appelées orbitales de spin . Pour un calcul d'orbitales atomiques , il s'agit généralement des orbitales d'un atome de type hydrogène (un atome avec un seul électron, mais la charge nucléaire appropriée). Pour un calcul d'orbitales moléculaires ou cristallines, les fonctions d'onde à un électron approximatives initiales sont généralement une combinaison linéaire d'orbitales atomiques (LCAO).
Les orbitales ci-dessus ne tiennent compte que de la présence d'autres électrons de manière moyenne. Dans la méthode Hartree-Fock, l'effet des autres électrons est pris en compte dans un contexte de théorie du champ moyen . Les orbitales sont optimisées en leur demandant de minimiser l'énergie du déterminant de Slater respectif. Les conditions variationnelles résultantes sur les orbitales conduisent à un nouvel opérateur à un électron, l' opérateur de Fock . Au minimum, les orbitales occupées sont des solutions propres de l'opérateur de Fock via une transformation unitaire entre elles. L'opérateur de Fock est un opérateur hamiltonien à un électron effectif étant la somme de deux termes. Le premier est une somme d'opérateurs d'énergie cinétique pour chaque électron, l'énergie de répulsion internucléaire et une somme de termes d'attraction coulombienne nucléaire-électronique . Le second est des termes de répulsion coulombienne entre électrons dans une description de la théorie du champ moyen ; une énergie de répulsion nette pour chaque électron du système, qui est calculée en traitant tous les autres électrons de la molécule comme une distribution lisse de charge négative. Il s’agit de la simplification majeure inhérente à la méthode Hartree-Fock et équivaut à la cinquième simplification de la liste ci-dessus.
Comme l'opérateur de Fock dépend des orbitales utilisées pour construire la matrice de Fock correspondante , les fonctions propres de l'opérateur de Fock sont à leur tour de nouvelles orbitales, qui peuvent être utilisées pour construire un nouvel opérateur de Fock. De cette manière, les orbitales Hartree-Fock sont optimisées de manière itérative jusqu'à ce que la variation de l'énergie électronique totale tombe en dessous d'un seuil prédéfini. De cette manière, un ensemble d'orbitales à un électron auto-cohérentes est calculé. La fonction d'onde électronique Hartree-Fock est alors le déterminant de Slater construit à partir de ces orbitales. En suivant les postulats de base de la mécanique quantique, la fonction d'onde Hartree-Fock peut ensuite être utilisée pour calculer n'importe quelle propriété chimique ou physique souhaitée dans le cadre de la méthode Hartree-Fock et des approximations utilisées.
Formulation mathématique
Dérivation
Selon les règles de Slater-Condon , la valeur moyenne de l'énergie de l' hamiltonien électronique moléculaire pour un déterminant de Slater est
où est l'opérateur à un électron incluant les opérateurs cinétiques électroniques et l'interaction coulombienne électron-noyau et
Pour dériver l'équation Hartree-Fock, nous minimisons la fonctionnelle énergétique pour N électrons avec des contraintes orthonormales.
Comme on peut choisir la base de , on choisit une base dans laquelle la matrice du multiplicateur de Lagrange devient diagonale, c'est-à-dire . En effectuant la variation , on obtient
Le facteur 1/2 dans l'hamiltonien moléculaire disparaît avant les intégrales doubles en raison de la symétrie et de la règle du produit. Nous pouvons définir l' opérateur de Fock pour réécrire l'équation
où l' opérateur de Coulomb et l' opérateur d'échange sont définis comme suit
L'opérateur d'échange n'a pas d'analogue classique et ne peut être défini que comme un opérateur intégral.
La solution et sont appelées respectivement orbitale moléculaire et énergie orbitale.
Bien que l'équation Hartree-Fock apparaisse sous la forme d'un problème de valeurs propres, l'opérateur Fock lui-même dépend et doit être résolu par une technique différente.
Énergie totale
L'énergie totale optimale peut être écrite en termes d'orbitales moléculaires.
Il convient de souligner que l’énergie totale n’est pas égale à la somme des énergies orbitales.
Si l'atome ou la molécule est à couche fermée , l'énergie totale selon la méthode Hartree-Fock est
Combinaison linéaire d'orbitales atomiques
En règle générale, dans les calculs Hartree–Fock modernes, les fonctions d'onde à un électron sont approximées par une combinaison linéaire d'orbitales atomiques . Ces orbitales atomiques sont appelées orbitales de type Slater . De plus, il est très courant que les « orbitales atomiques » utilisées soient en fait composées d'une combinaison linéaire d'une ou plusieurs orbitales de type gaussien , plutôt que d'orbitales de type Slater, dans le but d'économiser beaucoup de temps de calcul.
Différents ensembles de base sont utilisés dans la pratique, la plupart étant composés de fonctions gaussiennes. Dans certaines applications, une méthode d'orthogonalisation telle que le processus de Gram-Schmidt est effectuée afin de produire un ensemble de fonctions de base orthogonales. Cela peut en principe économiser du temps de calcul lorsque l'ordinateur résout les équations de Roothaan-Hall en convertissant efficacement la matrice de chevauchement en une matrice identité . Cependant, dans la plupart des programmes informatiques modernes pour les calculs moléculaires Hartree-Fock, cette procédure n'est pas suivie en raison du coût numérique élevé de l'orthogonalisation et de l'avènement d'algorithmes plus efficaces, souvent épars, pour résoudre le problème des valeurs propres généralisé , dont les équations de Roothaan-Hall sont un exemple.
Stabilité numérique
La stabilité numérique peut être un problème avec cette procédure et il existe plusieurs façons de lutter contre cette instabilité. L'une des plus basiques et généralement applicable est appelée F-mixage ou amortissement. Avec le F-mixage, une fois qu'une fonction d'onde d'un seul électron est calculée, elle n'est pas utilisée directement. Au lieu de cela, une combinaison de cette fonction d'onde calculée et des fonctions d'onde précédentes pour cet électron est utilisée, la plus courante étant une simple combinaison linéaire de la fonction d'onde calculée et de la fonction d'onde immédiatement précédente. Une astuce astucieuse, employée par Hartree, pour les calculs atomiques consistait à augmenter la charge nucléaire, rapprochant ainsi tous les électrons. Au fur et à mesure que le système se stabilisait, celle-ci était progressivement réduite à la charge correcte. Dans les calculs moléculaires, une approche similaire est parfois utilisée en calculant d'abord la fonction d'onde pour un ion positif, puis en utilisant ces orbitales comme point de départ pour la molécule neutre. Les programmes informatiques moléculaires modernes Hartree–Fock utilisent diverses méthodes pour assurer la convergence des équations de Roothaan–Hall.
Faiblesses, extensions et alternatives
Parmi les cinq simplifications décrites dans la section « Algorithme Hartree-Fock », la cinquième est généralement la plus importante. La négligence de la corrélation électronique peut conduire à de grands écarts par rapport aux résultats expérimentaux. Un certain nombre d'approches pour remédier à cette faiblesse, collectivement appelées méthodes post-Hartree-Fock , ont été conçues pour inclure la corrélation électronique à la fonction d'onde multiélectronique. L'une de ces approches, la théorie de la perturbation de Møller-Plesset , traite la corrélation comme une perturbation de l'opérateur de Fock. D'autres étendent la véritable fonction d'onde multiélectronique en termes de combinaison linéaire de déterminants de Slater, tels que le champ auto-cohérent multi-configurationnel , l'interaction de configuration , l'interaction de configuration quadratique et la SCF d'espace actif complet (CASSCF) . D'autres encore (comme la méthode de Monte Carlo quantique variationnelle ) modifient la fonction d'onde Hartree-Fock en la multipliant par une fonction de corrélation (facteur de « Jastrow »), un terme qui est explicitement une fonction de plusieurs électrons qui ne peut pas être décomposée en fonctions indépendantes à une seule particule.
Une alternative aux calculs Hartree–Fock utilisée dans certains cas est la théorie fonctionnelle de la densité , qui traite à la fois les énergies d'échange et de corrélation, bien qu'approximativement. En effet, il est courant d'utiliser des calculs qui sont un hybride des deux méthodes - le schéma populaire B3LYP est l'une de ces méthodes fonctionnelles hybrides . Une autre option consiste à utiliser des méthodes modernes de liaison de valence .
Progiciels
Pour une liste des logiciels connus pour gérer les calculs Hartree-Fock, en particulier pour les molécules et les solides, consultez la liste des logiciels de chimie quantique et de physique du solide .