La chimie computationnelle est une branche de la chimie qui utilise des simulations informatiques pour faciliter la résolution de problèmes chimiques. Elle emploie des méthodes de chimie théorique intégrées à des programmes informatiques pour calculer les structures et les propriétés des molécules , des groupes de molécules et des solides. Les chimistes computationnels s'attachent généralement à développer et à appliquer des programmes et des méthodologies informatiques à des questions chimiques spécifiques.
La complexité inhérente au problème à N corps accentue la difficulté de fournir des descriptions détaillées des systèmes quantiques. Les résultats de calculs peuvent compléter les informations obtenues par des expériences chimiques ou prédire des phénomènes chimiques non observés .
histoire de la mécanique quantique , les premiers calculs théoriques en chimie furent ceux de Walter Heitler et Fritz London en 1927, utilisant la théorie de la liaison de valence . Parmi les ouvrages qui ont influencé les débuts de la chimie quantique computationnelle, on peut citer Introduction to Quantum Mechanics – with Applications to Chemistry de Linus Pauling et E. Bright Wilson (1935) , Quantum Chemistry de Walter Eyring et Kimball (1944) , Elementary Wave Mechanics – with Applications to Quantum Chemistry de Heitler (1945) , et, plus tard , Valence de Coulson (1952) , qui ont tous constitué des références essentielles pour les chimistes au cours des décennies suivantes.Avec le développement de l'informatique performante dans les années 1940, la résolution d' équations d'ondes complexes pour les systèmes atomiques complexes devint un objectif réalisable. Au début des années 1950, les premiers calculs semi-empiriques d'orbitales atomiques furent effectués. Les chimistes théoriciens devinrent de grands utilisateurs des premiers ordinateurs numériques. Une avancée significative fut marquée par l'article de Clemens C.J. Roothaan paru en 1951 dans la revue Reviews of Modern Physics. Cet article portait principalement sur l'approche « LCAO MO » (combinaison linéaire d'orbitales atomiques et moléculaires). Smith et Sutcliffe en donnent une description très détaillée au Royaume-Uni. Les premiers calculs ab initio de la méthode Hartree-Fock sur des molécules diatomiques furent réalisés en 1956 au MIT, en utilisant un ensemble de base d' orbitales de Slater . Pour les molécules diatomiques, une étude systématique utilisant un ensemble de base minimal et le premier calcul avec un ensemble de base plus important ont été publiés respectivement par Ransil et Nesbet en 1960. Les premiers calculs polyatomiques utilisant des orbitales gaussiennes ont été effectués à la fin des années 1950. Les premiers calculs d'interaction de configuration ont été réalisés à Cambridge sur l' ordinateur EDSAC dans les années 1950 par Boys et ses collaborateurs, toujours avec des orbitales gaussiennes. En 1971, lors de la publication d'une bibliographie des calculs ab initio , les plus grandes molécules incluses étaient le naphtalène et l'azulène .
En 1964, des calculs par la méthode de Hückel (utilisant une méthode simple de combinaison linéaire d'orbitales atomiques (LCAO) pour déterminer les énergies électroniques des orbitales moléculaires des électrons π dans les systèmes hydrocarbonés conjugués) de molécules, dont la complexité varie du butadiène et du benzène à l'ovalène , ont été générés sur des ordinateurs à Berkeley et à Oxford. Ces méthodes empiriques ont été remplacées dans les années 1960 par des méthodes semi-empiriques telles que CNDO .
Au début des années 1970, des programmes informatiques ab initio performants tels qu'ATMOL, Gaussian , IBMOL et POLYAYTOM ont commencé à être utilisés pour accélérer les calculs ab initio des orbitales moléculaires. Parmi ces quatre programmes, seul Gaussian, aujourd'hui considérablement enrichi, est encore utilisé, mais de nombreux autres programmes le sont désormais. Parallèlement, les méthodes de la mécanique moléculaire , telles que le champ de force MM2 , ont été développées, principalement par Norman Allinger .
L'une des premières mentions du terme « chimie computationnelle » se trouve dans l'ouvrage de 1970 intitulé *Computers and Their Role in the Physical Sciences*, de Sidney Fernbach et Abraham Haskell Taub, où ils affirment : « Il semble donc que la chimie computationnelle puisse enfin devenir une réalité de plus en plus concrète. » Au cours des années 1970, des méthodes très diverses ont commencé à être perçues comme faisant partie d'une nouvelle discipline émergente : la chimie computationnelle . Le *Journal of Computational Chemistry* a été publié pour la première fois en 1980.
La chimie computationnelle a été récompensée par plusieurs prix Nobel. En 1998, le prix Nobel de chimie a été décerné à Walter Kohn , « pour ses travaux sur la théorie de la fonctionnelle de la densité », et à John Pople , « pour ses travaux sur les méthodes de calcul en chimie quantique » . Martin Karplus , Michael Levitt et Arieh Warshel ont reçu le prix Nobel de chimie en 2013 pour « le développement de modèles multi-échelles pour les systèmes chimiques complexes »
Applications
La chimie computationnelle a un large éventail d'applications
- La prédiction de la structure moléculaire des molécules par l'utilisation de la simulation des forces, ou de méthodes de chimie quantique plus précises, pour trouver des points stationnaires sur la surface d'énergie lorsque la position des noyaux est modifiée.
- Stockage et recherche de données sur les entités chimiques (voir bases de données chimiques ).
- Identification des corrélations entre les structures chimiques et les propriétés (voir relation quantitative structure-propriété (QSPR) et relation quantitative structure-activité (QSAR)).
- Approches informatiques pour faciliter la synthèse efficace de composés.
- Approches informatiques pour concevoir des molécules qui interagissent de manière spécifique avec d'autres molécules (par exemple, la conception de médicaments et la catalyse .
Ces domaines peuvent donner lieu à plusieurs applications, comme indiqué ci-dessous.
Catalyse
La chimie computationnelle est un outil d'analyse des systèmes catalytiques qui permet de réaliser des expériences. La théorie moderne de la structure électronique et la théorie de la fonctionnelle de la densité ont permis aux chercheurs de découvrir et de comprendre les catalyseurs . Les études computationnelles appliquent la chimie théorique à la recherche en catalyse. Les méthodes de la théorie de la fonctionnelle de la densité calculent les énergies et les orbitales des molécules afin de modéliser leurs structures . Grâce à ces méthodes, les chercheurs peuvent prédire des valeurs telles que l'énergie d'activation , la réactivité des sites .
Les données difficiles à obtenir expérimentalement peuvent être obtenues grâce à des méthodes de calcul permettant de modéliser les mécanismes des cycles catalytiques. Des chimistes computationnels expérimentés fournissent des prédictions proches des données expérimentales, en tenant compte des méthodes et des ensembles de bases appropriés. Grâce à des données de calcul de qualité, les chercheurs peuvent prédire comment améliorer les catalyseurs afin de réduire les coûts et d'accroître l'efficacité de ces réactions.
Développement de médicaments
La chimie computationnelle est utilisée dans le développement de médicaments pour modéliser des molécules potentiellement utiles et aider les entreprises à gagner du temps et à réduire les coûts. Le processus de découverte de médicaments comprend l'analyse de données, la recherche de moyens d'améliorer les molécules existantes, l'identification de voies de synthèse et l'évaluation de ces molécules. La chimie computationnelle facilite ce processus en prédisant les expériences les plus pertinentes à réaliser, évitant ainsi d'en mener d'autres. Les méthodes de calcul permettent également de déterminer des valeurs difficiles à obtenir expérimentalement, comme le pKa des composés. Des méthodes telles que la théorie de la fonctionnelle de la densité (DFT) peuvent être utilisées pour modéliser les molécules médicamenteuses et déterminer leurs propriétés, comme les énergies de leurs orbitales HOMO et LUMO et leurs orbitales moléculaires.
Outre la synthèse de médicaments, les vecteurs de médicaments font également l'objet de recherches par les chimistes computationnels pour les nanomatériaux . Cela permet aux chercheurs de simuler des environnements afin de tester l'efficacité et la stabilité des vecteurs de médicaments. Comprendre comment l'eau interagit avec ces nanomatériaux garantit la stabilité du matériau dans le corps humain. Ces simulations informatiques aident les chercheurs à optimiser le matériau et à trouver la meilleure façon de structurer ces nanomatériaux avant leur fabrication.
bases de données de chimie computationnelle
Méthodes
méthode ab initio
Les méthodes ab initio nécessitent la définition d'un niveau de théorie (la méthode) et d'un ensemble de base. Un ensemble de base est constitué de fonctions centrées sur les atomes de la molécule. Ces ensembles sont ensuite utilisés pour décrire les orbitales moléculaires via l' ansatz de la méthode des orbitales moléculaires par combinaison linéaire d'orbitales atomiques (LCAO) .
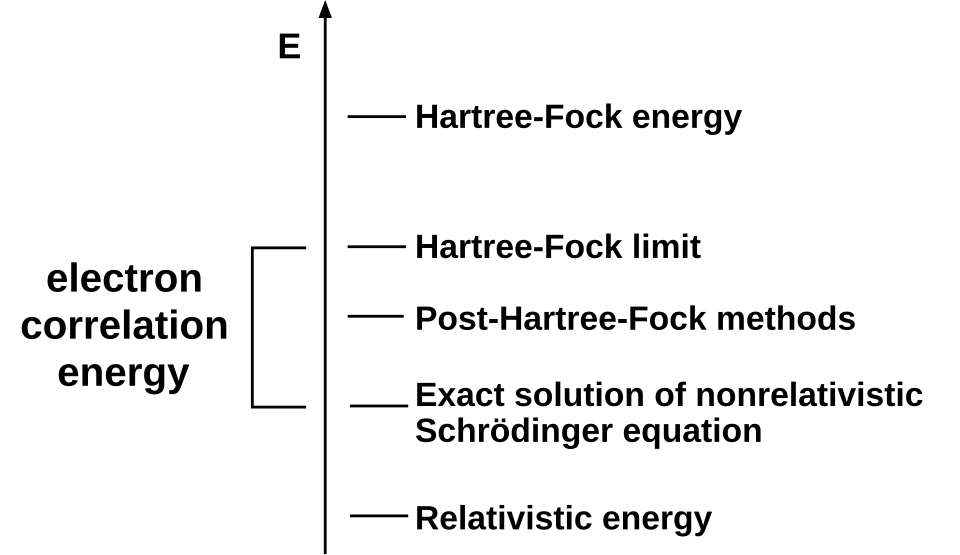
Un type courant de calcul ab initio de la structure électronique est la méthode Hartree-Fock (HF), une extension de la théorie des orbitales moléculaires , où les répulsions électron-électron dans la molécule ne sont pas prises en compte spécifiquement ; seul l’effet moyen des électrons est inclus dans le calcul. Lorsque la taille de la base augmente, l’énergie et la fonction d’onde tendent vers une limite appelée limite Hartree-Fock.
De nombreux calculs débutent par un calcul Hartree-Fock, puis corrigent la répulsion électron-électron, également appelée corrélation électronique . Ces calculs sont dits post-Hartree-Fock . En améliorant continuellement ces méthodes, les scientifiques peuvent se rapprocher de plus en plus d'une prédiction parfaite du comportement des systèmes atomiques et moléculaires dans le cadre de la mécanique quantique, telle que définie par l'équation de Schrödinger.
Dans la plupart des cas, la fonction d'onde Hartree-Fock occupe une seule configuration ou un seul déterminant. Dans certains cas, notamment pour les processus de rupture de liaison, cela est insuffisant et plusieurs configurations doivent être utilisées.
L'énergie moléculaire totale peut être évaluée en fonction de la géométrie moléculaire ; autrement dit, de la surface d'énergie potentielle . Une telle surface peut être utilisée pour la dynamique des réactions. Les points stationnaires de la surface permettent de prédire les différents isomères et les structures de transition pour la conversion entre isomères.
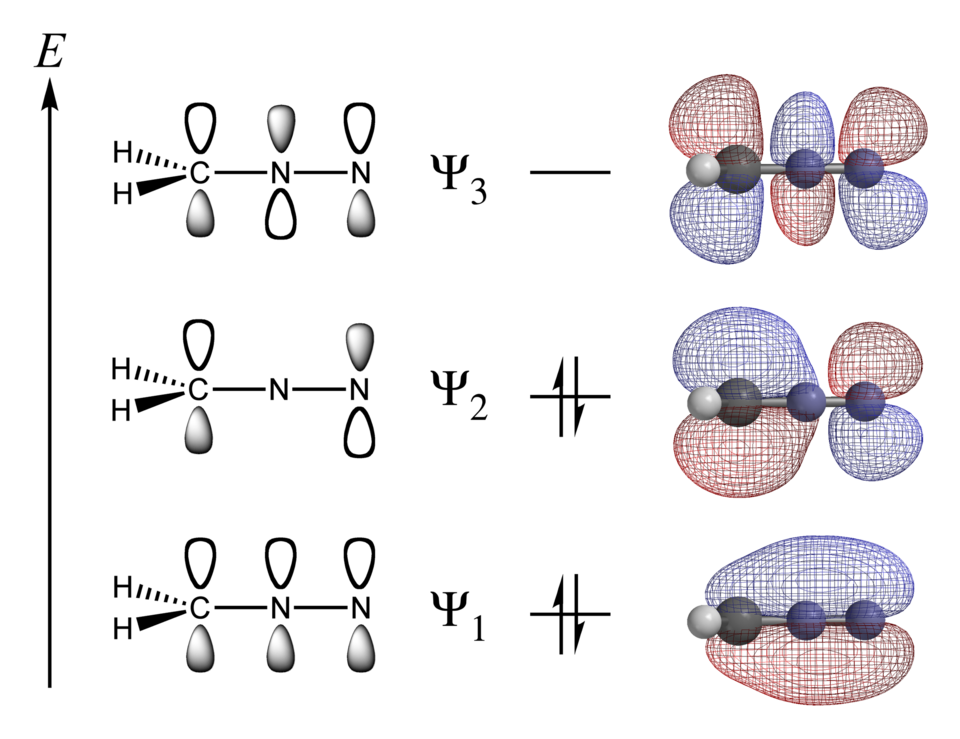
thermochimie computationnelle
Dynamique chimique
Après séparation des variables électroniques et nucléaires dans la représentation de Born-Oppenheimer, le paquet d'ondes correspondant aux degrés de liberté nucléaires est propagé via l' opérateur d'évolution temporelle (physique) associé à l' équation de Schrödinger dépendante du temps (pour l' hamiltonien moléculaire complet ). Dans l' approche complémentaire dépendante de l'énergie, l' équation de Schrödinger indépendante du temps est résolue à l'aide du formalisme de la théorie de la diffusion . Le potentiel représentant l'interaction interatomique est donné par les surfaces d'énergie potentielle . En général, ces surfaces sont couplées par des termes de couplage vibronique .
Les méthodes les plus courantes pour propager le paquet d'ondes associé à la géométrie moléculaire sont :
- le polynôme de Tchebychev (réel) ,
- la méthode Hartree dépendante du temps multiconfiguration (MCTDH),
- la méthode semi-classique
- et la technique de l'opérateur divisé expliquée ci-dessous.
technique de l'opérateur divisé
La manière dont une méthode de calcul résout les équations quantiques influence sa précision et son efficacité. La technique de l'opérateur fractionné est l'une de ces méthodes de résolution d'équations différentielles. En chimie computationnelle, cette technique réduit les coûts de calcul liés à la simulation de systèmes chimiques. Ces coûts correspondent au temps nécessaire aux ordinateurs pour calculer ces systèmes, ce temps pouvant atteindre plusieurs jours pour les systèmes complexes. La résolution des systèmes quantiques est difficile et chronophage pour l'humain. Les méthodes de l'opérateur fractionné permettent aux ordinateurs de calculer rapidement ces systèmes en résolvant les sous-problèmes d'une équation différentielle quantique . Cette méthode consiste à séparer l'équation différentielle en deux équations distinctes, notamment lorsqu'il y a plus de deux opérateurs. Une fois résolues, les équations fractionnées sont combinées en une seule équation pour obtenir une solution plus facilement calculable.
Cette méthode est utilisée dans de nombreux domaines nécessitant la résolution d'équations différentielles, comme la biologie . Cependant, elle présente une erreur de séparation. Par exemple, avec la solution suivante pour une équation différentielle :
L’équation peut être décomposée, mais les solutions ne seront pas exactes, seulement similaires. Il s’agit d’un exemple de décomposition du premier ordre.
Il existe des moyens de réduire cette erreur, notamment en prenant la moyenne de deux équations fractionnées.
Une autre façon d'améliorer la précision consiste à utiliser un fractionnement d'ordre supérieur. Généralement, le fractionnement d'ordre deux est le maximum utilisé, car les fractionnements d'ordre supérieur nécessitent un temps de calcul beaucoup plus long et leur coût est disproportionné. Les méthodes d'ordre supérieur deviennent trop complexes à mettre en œuvre et, malgré leur précision accrue, ne sont pas utiles pour la résolution d'équations différentielles.
Les chimistes computationnels consacrent beaucoup de temps à améliorer la précision des systèmes calculés à l'aide de la technique de l'opérateur fractionné tout en minimisant le coût de calcul. Les méthodes de calcul représentent un défi majeur pour de nombreux chimistes qui tentent de simuler des molécules ou des environnements chimiques.
Méthodes de la fonctionnelle de la densité
Méthodes semi-empiriques
Des méthodes semi-empiriques primitives ont été conçues antérieurement, dans lesquelles la partie à deux électrons de l' hamiltonien n'est pas explicitement incluse. Pour les systèmes à électrons π, il s'agissait de la méthode de Hückel proposée par Erich Hückel , et pour tous les systèmes à électrons de valence, de la méthode de Hückel étendue proposée par Roald Hoffmann . Les méthodes de Hückel sont parfois qualifiées de « complètement empiriques » car elles ne reposent pas sur un hamiltonien. Cependant, le terme « méthodes empiriques » ou « champs de force empiriques » est généralement utilisé pour décrire la mécanique moléculaire.
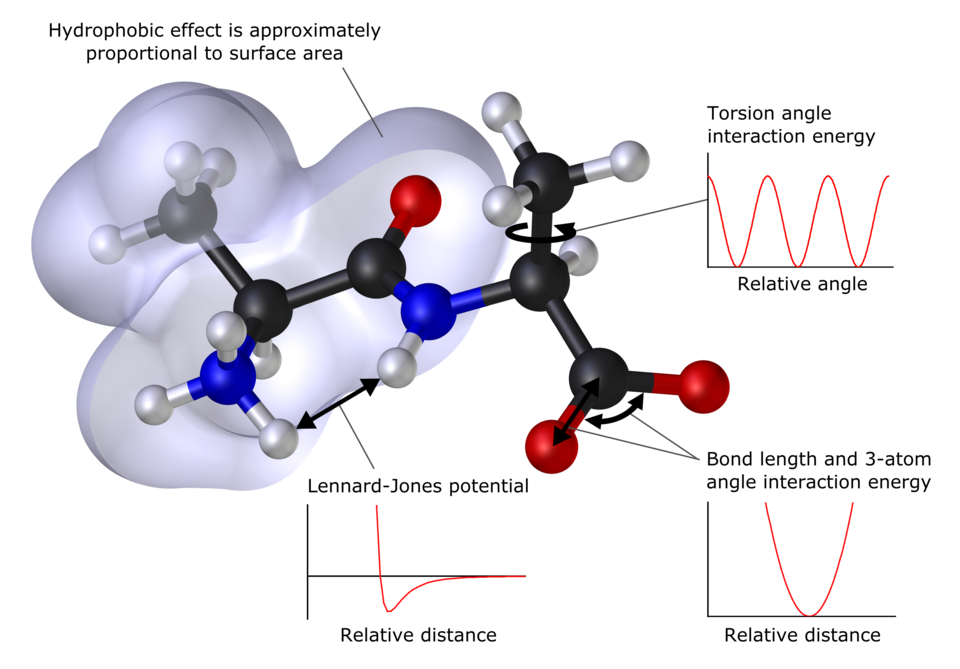
Mécanique moléculaire
La base de données de composés utilisée pour la paramétrisation, c'est-à-dire l'ensemble résultant de paramètres et de fonctions, appelé champ de force , est essentielle à la réussite des calculs de mécanique moléculaire. Un champ de force paramétré pour une classe spécifique de molécules, par exemple les protéines, ne sera pertinent que pour la description d'autres molécules de la même classe. Ces méthodes peuvent être appliquées aux protéines et à d'autres grandes molécules biologiques, et permettent d'étudier l'approche et l'interaction (amarrage) de molécules médicamenteuses potentielles.
Dynamique moléculaire
Monte Carlo
La méthode de Monte Carlo (MC) génère des configurations d'un système en modifiant aléatoirement la position de ses particules, ainsi que leur orientation et leur conformation, le cas échéant. Il s'agit d'une méthode d'échantillonnage aléatoire qui utilise l' échantillonnage d'importance . Les méthodes d'échantillonnage d'importance permettent de générer des états de basse énergie, ce qui autorise un calcul précis des propriétés. L'énergie potentielle de chaque configuration du système peut être calculée, ainsi que les valeurs d'autres propriétés, à partir des positions des atomes.
Mécanique quantique/mécanique moléculaire (MQ/MM)
Chimie quantique computationnelle
Les méthodes conventionnelles de chimie computationnelle peinent souvent à résoudre les équations complexes de la mécanique quantique, notamment en raison de la croissance exponentielle de la fonction d'onde d'un système quantique. La chimie computationnelle quantique relève ces défis grâce à des méthodes de calcul quantique , telles que la qubitisation et l'estimation de phase quantique , qui sont censées offrir des solutions évolutives.
La qubitisation consiste à adapter l'opérateur hamiltonien pour un traitement plus efficace sur les ordinateurs quantiques, améliorant ainsi l'efficacité de la simulation. L'estimation de la phase quantique, quant à elle, permet de déterminer avec précision les états propres de l'énergie, essentiels à la compréhension du comportement du système quantique.
Bien que ces techniques aient fait progresser le domaine de la chimie computationnelle, notamment la simulation des systèmes chimiques, leur application pratique reste actuellement limitée aux systèmes de petite taille en raison de contraintes technologiques. Néanmoins, ces développements pourraient permettre des avancées significatives vers des simulations de chimie quantique plus précises et plus économes en ressources.
Coûts de calcul des algorithmes de chimie
Exemples de complexité algorithmique
La liste suivante illustre l'impact de la complexité de calcul sur les algorithmes utilisés en chimie. Bien qu'elle fournisse des exemples clés, elle n'est pas exhaustive et sert de guide pour comprendre comment les exigences de calcul influencent le choix des méthodes de calcul spécifiques en chimie.
Dynamique moléculaire
Mécanique quantique/mécanique moléculaire (MQ/MM)
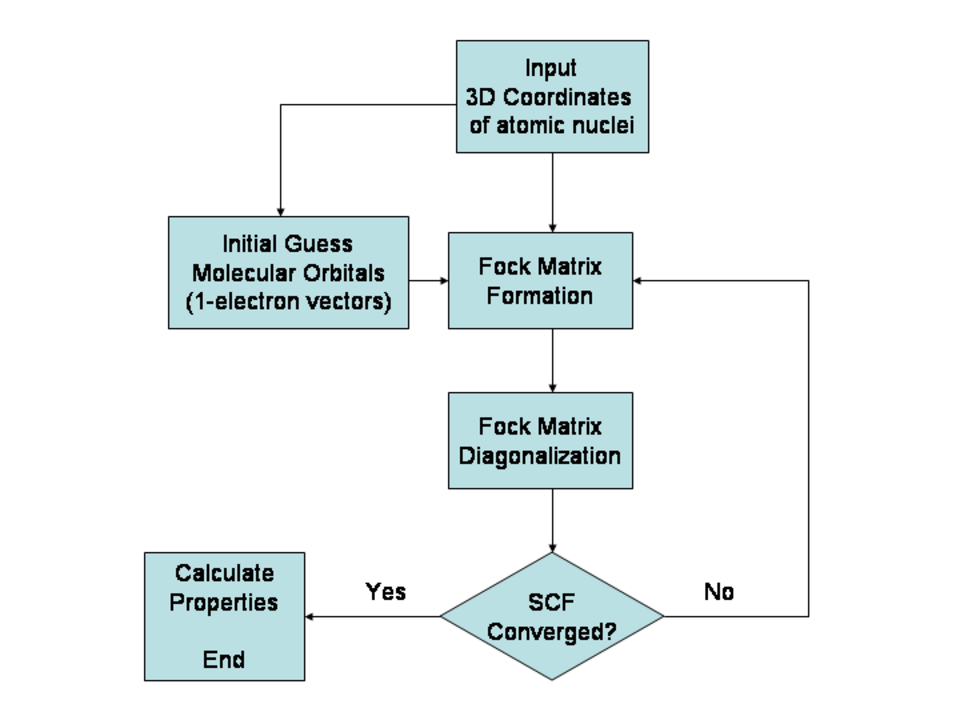
Méthode Hartree-Fock
Théorie de la fonctionnelle de la densité
Complexité
Les implémentations traditionnelles de la DFT présentent généralement une complexité de l'ordre de O(n) , principalement en raison de la nécessité de diagonaliser la matrice de Kohn-Sham . L'étape de diagonalisation, qui détermine les valeurs propres et les vecteurs propres de la matrice, contribue le plus à cette complexité. Les avancées récentes en DFT visent à réduire cette complexité grâce à diverses approximations et améliorations algorithmiques . Des gains de vitesse significatifs ont également été observés dans les solveurs de valeurs propres, en particulier le solveur ELPA , utilisé dans de nombreux codes
La DFT a une complexité QMA .
Méthode CCSD et CCSD(T) standard

Méthode CCSD(T) à mise à l'échelle linéaire
La précision peut toujours être améliorée au prix d'un coût de calcul plus élevé. Des erreurs importantes peuvent apparaître dans les modèles ab initio comportant un grand nombre d'électrons, en raison du coût de calcul des méthodes relativistes complètes. Ceci complique l'étude des molécules interagissant avec des atomes de masse atomique élevée, tels que les métaux de transition et leurs propriétés catalytiques. Les algorithmes actuels de chimie computationnelle permettent de calculer couramment les propriétés de petites molécules contenant jusqu'à une quarantaine d'électrons, avec des erreurs inférieures à quelques kJ/mol pour les énergies. Concernant les géométries, les longueurs de liaison peuvent être prédites à quelques picomètres près et les angles de liaison à 0,5 degré près. Le traitement de molécules plus grandes, contenant quelques dizaines d'atomes, est réalisable par des méthodes plus approchées telles que la théorie de la fonctionnelle de la densité (DFT).
Il existe un débat au sein du domaine quant à la suffisance de ces dernières méthodes pour décrire des réactions chimiques complexes, telles que celles rencontrées en biochimie. Les grosses molécules peuvent être étudiées par des méthodes semi-empiriques approchées. Les molécules encore plus grandes sont traitées par des méthodes de mécanique classique utilisant ce que l'on appelle la mécanique moléculaire (MM). Dans les méthodes QM-MM, de petites parties de grands complexes sont traitées de manière quantique (QM), et le reste est traité de manière approchée (MM).
Logiciels
Il existe de nombreux logiciels de chimie computationnelle autonomes . Certains proposent un large éventail de méthodes, tandis que d'autres se concentrent sur un domaine très spécifique, voire sur une seule méthode. Vous trouverez des informations détaillées sur la plupart d'entre eux dans :
- Programmes de modélisation biomoléculaire : protéines , acides nucléiques .
- Programmes de mécanique moléculaire .
- Logiciels de chimie quantique et de physique du solide prenant en charge plusieurs méthodes.
- Logiciel de conception moléculaire
- Programmes semi-empiriques .
- Programmes de liaison de valence .
Revues spécialisées en chimie computationnelle
- Rapports annuels en chimie computationnelle
- Chimie computationnelle et théorique
- Science computationnelle et théorique des polymères
- Informatique et génie chimique
- Journal d'information et de modélisation chimique
- Journal des logiciels chimiques
- Journal de théorie chimique et de calcul
- Journal of Cheminformatics
- Journal de chimie computationnelle
- Journal de chimie assistée par ordinateur
- Journal de chimie informatique du Japon
- Journal de conception moléculaire assistée par ordinateur
- Journal de chimie théorique et computationnelle
- Informatique moléculaire
- Comptes rendus de chimie théorique
Pour en savoir plus
- Schaefer, Henry F. III (1984). Chimie quantique . Oxford : Clarendon Press. Résumés de nombreux développements antérieurs de la théorie ab initio .