En biologie moléculaire , un domaine protéique est une région de la chaîne polypeptidique d' une protéine qui est auto-stabilisante et qui se replie indépendamment du reste. Chaque domaine forme une structure tridimensionnelle compacte et pliée . De nombreuses protéines sont constituées de plusieurs domaines, et un domaine peut apparaître dans une variété de protéines différentes. L'évolution moléculaire utilise les domaines comme blocs de construction et ceux-ci peuvent être recombinés dans différents arrangements pour créer des protéines avec différentes fonctions. En général, les domaines varient en longueur d'environ 50 acides aminés jusqu'à 250 acides aminés de longueur. Les domaines les plus courts, tels que les doigts de zinc , sont stabilisés par des ions métalliques ou des ponts disulfures . Les domaines forment souvent des unités fonctionnelles, telles que le domaine de la main EF de liaison au calcium de la calmoduline . Parce qu'ils sont indépendamment stables, les domaines peuvent être « échangés » par génie génétique entre une protéine et une autre pour fabriquer des protéines chimériques .
Arrière-plan
Le concept de domaine a été proposé pour la première fois en 1973 par Wetlaufer après des études de cristallographie aux rayons X du lysozyme de poule et de la papaïne et par des études de protéolyse limitées des immunoglobulines . Wetlaufer a défini les domaines comme des unités stables de structure protéique qui pouvaient se replier de manière autonome. Dans le passé, les domaines ont été décrits comme des unités de :
- structure compacte
- fonction et évolution
- pliage.
Chaque définition est valable et se chevauchera souvent, c'est-à-dire qu'un domaine structurel compact qui se trouve parmi diverses protéines est susceptible de se replier indépendamment dans son environnement structurel. La nature rassemble souvent plusieurs domaines pour former des protéines multidomaines et multifonctionnelles offrant un grand nombre de possibilités. Dans une protéine multidomaine, chaque domaine peut remplir sa propre fonction de manière indépendante ou de manière concertée avec ses voisins. Les domaines peuvent soit servir de modules pour construire de grands assemblages tels que des particules virales ou des fibres musculaires, soit fournir des sites catalytiques ou de liaison spécifiques comme ceux que l'on trouve dans les enzymes ou les protéines régulatrices.
Exemple : Pyruvate kinase
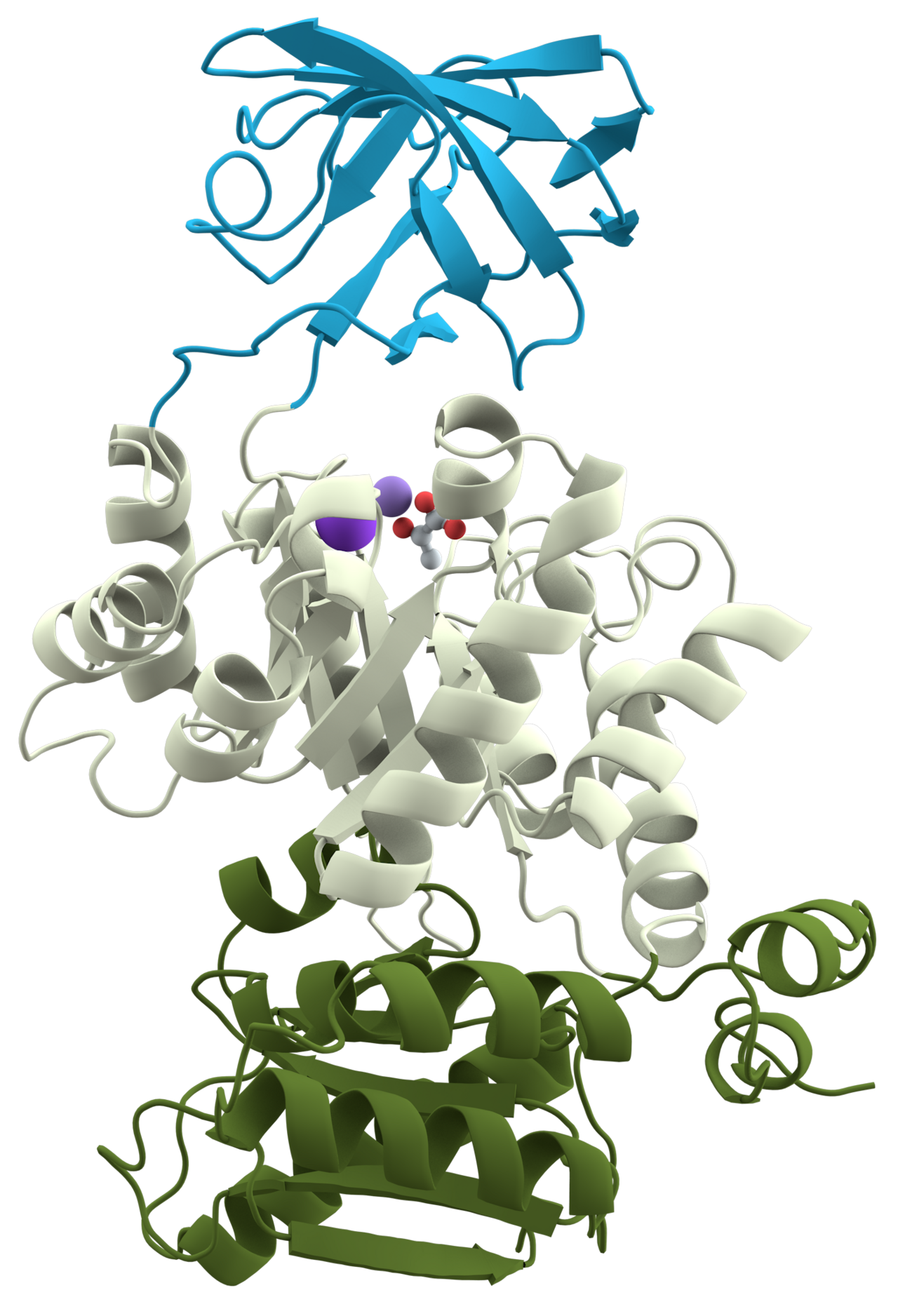
Un exemple approprié est la pyruvate kinase (voir la première figure), une enzyme glycolytique qui joue un rôle important dans la régulation du flux du fructose-1,6-biphosphate au pyruvate. Elle contient un domaine de liaison aux nucléotides β (en bleu), un domaine de liaison au substrat α/β (en gris) et un domaine de régulation α/β (en vert olive), reliés par plusieurs lieurs polypeptidiques. Chaque domaine de cette protéine apparaît dans divers ensembles de familles de protéines .
Le domaine de liaison du substrat du tonneau α/β central est l'un des replis enzymatiques les plus courants . On le retrouve dans de nombreuses familles d'enzymes différentes, catalysant des réactions totalement indépendantes. Le tonneau α/β est communément appelé le tonneau TIM, du nom de la triose phosphate isomérase, qui a été la première structure de ce type à être résolue. Il est actuellement classé en 26 familles homologues dans la base de données des domaines CATH. Le tonneau TIM est formé d'une séquence de motifs β-α-β fermés par la liaison hydrogène du premier et du dernier brin, formant un tonneau à huit brins. Il existe un débat sur l'origine évolutive de ce domaine. Une étude a suggéré qu'une seule enzyme ancestrale aurait pu diverger en plusieurs familles, tandis qu'une autre suggère qu'une structure stable de tonneau TIM a évolué par évolution convergente.
Le tonneau TIM de la pyruvate kinase est « discontinu », ce qui signifie que plus d'un segment du polypeptide est nécessaire pour former le domaine. Cela est probablement le résultat de l'insertion d'un domaine dans un autre au cours de l'évolution de la protéine. Il a été démontré à partir de structures connues qu'environ un quart des domaines structurels sont discontinus. Le domaine régulateur du tonneau β inséré est « continu », constitué d'un seul segment de polypeptide.
Unités de structure des protéines
La structure primaire (chaîne d'acides aminés) d'une protéine code finalement sa conformation tridimensionnelle (3D) pliée de manière unique. Le facteur le plus important qui régit le repliement d'une protéine en structure 3D est la distribution des chaînes latérales polaires et non polaires. Le repliement est provoqué par l'enfouissement des chaînes latérales hydrophobes à l'intérieur de la molécule afin d'éviter tout contact avec l'environnement aqueux. En général, les protéines ont un noyau de résidus hydrophobes entouré d'une coque de résidus hydrophiles. Étant donné que les liaisons peptidiques elles-mêmes sont polaires, elles sont neutralisées par des liaisons hydrogène entre elles lorsqu'elles se trouvent dans l'environnement hydrophobe. Cela donne naissance à des régions du polypeptide qui forment des motifs structurels 3D réguliers appelés structure secondaire . Il existe deux principaux types de structure secondaire : les hélices α et les feuillets β .
Certaines combinaisons simples d'éléments de structure secondaire se produisent fréquemment dans la structure des protéines et sont appelées structures ou motifs supersecondaires . Par exemple, le motif en épingle à cheveux β se compose de deux brins β antiparallèles adjacents reliés par une petite boucle. Il est présent dans la plupart des structures β antiparallèles à la fois sous forme de ruban isolé et dans le cadre de feuillets β plus complexes. Une autre structure supersecondaire courante est le motif β-α-β, qui est fréquemment utilisé pour connecter deux brins β parallèles. L'hélice α centrale relie les extrémités C du premier brin aux extrémités N du deuxième brin, plaquant ses chaînes latérales contre le feuillet β et protégeant ainsi les résidus hydrophobes des brins β de la surface.
L'association covalente de deux domaines représente un avantage fonctionnel et structurel puisqu'elle augmente la stabilité par rapport aux mêmes structures associées de manière non covalente. D'autres avantages sont la protection des intermédiaires dans les fentes enzymatiques inter-domaines qui pourraient autrement être instables dans des environnements aqueux, et un rapport stoechiométrique fixe de l'activité enzymatique nécessaire pour un ensemble séquentiel de réactions.
L'alignement structurel est un outil important pour déterminer les domaines.
Structure tertiaire
Plusieurs motifs se regroupent pour former des unités compactes, locales et semi-indépendantes appelées domaines. La structure 3D globale de la chaîne polypeptidique est appelée structure tertiaire de la protéine . Les domaines sont les unités fondamentales de la structure tertiaire, chaque domaine contenant un noyau hydrophobe individuel construit à partir d'unités structurelles secondaires reliées par des régions en boucle. L'emballage du polypeptide est généralement beaucoup plus serré à l'intérieur qu'à l'extérieur du domaine, produisant un noyau solide et une surface fluide. Les résidus de noyau sont souvent conservés dans une famille de protéines , tandis que les résidus dans les boucles sont moins conservés, à moins qu'ils ne soient impliqués dans la fonction de la protéine. La structure tertiaire des protéines peut être divisée en quatre classes principales en fonction du contenu structurel secondaire du domaine.
- Tous les domaines alpha ont un noyau constitué exclusivement d'hélices alpha. Cette classe est dominée par de petits replis, dont beaucoup forment un faisceau simple avec des hélices montantes et descendantes.
- Tous les domaines β ont un noyau composé de feuillets β antiparallèles, généralement deux feuillets empilés l'un contre l'autre. Divers motifs peuvent être identifiés dans la disposition des brins, donnant souvent lieu à l'identification de motifs récurrents, par exemple le motif de clé grecque.
- Les domaines α+β sont un mélange de motifs entièrement α et entièrement β. La classification des protéines dans cette classe est difficile en raison des chevauchements avec les trois autres classes et n'est donc pas utilisée dans la base de données des domaines CATH .
- Les domaines α/β sont constitués d'une combinaison de motifs β-α-β qui forment principalement une feuille β parallèle entourée d'hélices α amphipathiques. Les structures secondaires sont disposées en couches ou en barils.
Limites de taille
Les domaines ont des limites de taille. La taille des domaines structurels individuels varie de 36 résidus dans la E-sélectine à 692 résidus dans la lipoxygénase-1, mais la majorité, 90 %, ont moins de 200 résidus avec une moyenne d'environ 100 résidus. Les domaines très courts, moins de 40 résidus, sont souvent stabilisés par des ions métalliques ou des liaisons disulfures. Les domaines plus grands, supérieurs à 300 résidus, sont susceptibles d'être constitués de plusieurs noyaux hydrophobes.
Structure quaternaire
De nombreuses protéines ont une structure quaternaire , qui se compose de plusieurs chaînes polypeptidiques qui s'associent pour former une molécule oligomérique. Chaque chaîne polypeptidique d'une telle protéine est appelée sous-unité. L'hémoglobine, par exemple, se compose de deux sous-unités α et de deux sous-unités β. Chacune des quatre chaînes possède un repli entièrement constitué de globine α avec une poche hémique.
Échange de domaine
L'échange de domaines est un mécanisme de formation d'assemblages oligomériques. Dans l'échange de domaines, un élément secondaire ou tertiaire d'une protéine monomérique est remplacé par le même élément d'une autre protéine. L'échange de domaines peut aller d'éléments de structure secondaires à des domaines structuraux entiers. Il représente également un modèle d'évolution pour l'adaptation fonctionnelle par oligomérisation, par exemple les enzymes oligomériques dont le site actif se situe aux interfaces des sous-unités.
Les domaines comme modules évolutifs
La nature est une bricoleuse et non une inventeuse , les nouvelles séquences sont adaptées à partir de séquences préexistantes plutôt qu'inventées. Les domaines sont le matériel commun utilisé par la nature pour générer de nouvelles séquences ; ils peuvent être considérés comme des unités génétiquement mobiles, appelées « modules ». Souvent, les extrémités C et N des domaines sont proches l'une de l'autre dans l'espace, ce qui leur permet d'être facilement « insérées » dans les structures parentales au cours du processus d'évolution. De nombreuses familles de domaines se trouvent dans les trois formes de vie, les archées , les bactéries et les eucaryotes . Les modules protéiques sont un sous-ensemble de domaines protéiques que l'on trouve dans une gamme de protéines différentes avec une structure particulièrement polyvalente. On en trouve des exemples parmi les protéines extracellulaires associées à la coagulation, à la fibrinolyse, au complément, à la matrice extracellulaire, aux molécules d'adhésion à la surface cellulaire et aux récepteurs de cytokines. Quatre exemples concrets de modules protéiques répandus sont les domaines suivants : SH2 , immunoglobuline , fibronectine de type 3 et le kringle .
L'évolution moléculaire donne naissance à des familles de protéines apparentées ayant une séquence et une structure similaires. Cependant, les similitudes de séquence peuvent être extrêmement faibles entre des protéines qui partagent la même structure. Les structures protéiques peuvent être similaires parce que les protéines ont divergé d'un ancêtre commun. Alternativement, certains replis peuvent être plus favorisés que d'autres car ils représentent des arrangements stables de structures secondaires et certaines protéines peuvent converger vers ces replis au cours de l'évolution. Il existe actuellement environ 110 000 structures 3D de protéines déterminées expérimentalement déposées dans la Protein Data Bank (PDB). Cependant, cet ensemble contient de nombreuses structures identiques ou très similaires. Toutes les protéines doivent être classées en familles structurelles pour comprendre leurs relations évolutives. Les comparaisons structurelles sont mieux réalisées au niveau du domaine. Pour cette raison, de nombreux algorithmes ont été développés pour attribuer automatiquement des domaines dans les protéines ayant une structure 3D connue (voir § Définition de domaine à partir de coordonnées structurelles).
La base de données de domaines CATH classe les domaines en environ 800 familles de replis ; dix de ces replis sont très peuplés et sont appelés « super-plis ». Les super-plis sont définis comme des plis pour lesquels il existe au moins trois structures sans similarité de séquence significative. Le plus peuplé est le super-pli en tonneau α/β, comme décrit précédemment.
Protéines multidomaines
La majorité des protéines, les deux tiers chez les organismes unicellulaires et plus de 80 % chez les métazoaires, sont des protéines multidomaines. Cependant, d'autres études ont conclu que 40 % des protéines procaryotes sont constituées de plusieurs domaines tandis que les eucaryotes ont environ 65 % de protéines multidomaines.
De nombreux domaines dans les protéines multidomaines eucaryotes peuvent être trouvés comme protéines indépendantes chez les procaryotes, suggérant que des domaines dans des protéines multidomaines ont existé autrefois comme protéines indépendantes. Par exemple, les vertébrés ont un polypeptide multi-enzyme contenant les domaines GAR synthétase , AIR synthétase et GAR transformylase (GARs-AIRs-GARt ; GAR : glycinamide ribonucleotide synthetase/transferase ; AIR : aminoimidazole ribonucleotide synthetase). Chez les insectes, le polypeptide apparaît sous la forme GARs-(AIRs)2-GARt, chez la levure GARs-AIRs est codé séparément de GARt, et chez les bactéries chaque domaine est codé séparément.

Origine
Les protéines multidomaines sont probablement apparues sous l'effet d'une pression sélective exercée au cours de l'évolution pour créer de nouvelles fonctions. Diverses protéines ont divergé de leurs ancêtres communs par différentes combinaisons et associations de domaines. Les unités modulaires se déplacent fréquemment, au sein et entre les systèmes biologiques, par des mécanismes de brassage génétique :
- transposition d'éléments mobiles incluant les transferts horizontaux (entre espèces) ;
- réarrangements bruts tels que inversions, translocations, suppressions et duplications ;
- recombinaison homologue ;
- glissement de l'ADN polymérase lors de la réplication.
Types d'organisation
L'organisation multidomaine la plus simple observée dans les protéines est celle d'un seul domaine répété en tandem. Les domaines peuvent interagir les uns avec les autres ( interaction domaine-domaine ) ou rester isolés, comme des perles sur un fil. La protéine musculaire géante titine de 30 000 résidus comprend environ 120 domaines de type fibronectine-III et de type Ig. Dans les protéases à sérine, un événement de duplication de gène a conduit à la formation d'une enzyme à deux domaines à barillet β. Les répétitions ont divergé si largement qu'il n'y a pas de similitude de séquence évidente entre elles. Le site actif est situé dans une fente entre les deux domaines à barillet β, dans laquelle des résidus fonctionnellement importants sont apportés par chaque domaine. Des mutants génétiquement modifiés de la protéase à sérine chymotrypsine se sont avérés avoir une certaine activité protéinase même si leurs résidus de site actif ont été abolis et il a donc été postulé que l'événement de duplication a amélioré l'activité de l'enzyme.
Les modules présentent souvent des relations de connectivité différentes, comme l'illustrent les kinésines et les transporteurs ABC . Le domaine moteur de la kinésine peut se trouver à l'une ou l'autre extrémité d'une chaîne polypeptidique qui comprend une région en spirale et un domaine de chargement. Les transporteurs ABC sont construits avec jusqu'à quatre domaines constitués de deux modules non apparentés, une cassette de liaison à l'ATP et un module membranaire intégral, disposés selon diverses combinaisons.
Non seulement les domaines se recombinent, mais il existe de nombreux exemples d'un domaine inséré dans un autre. Les similitudes de séquence ou de structure avec d'autres domaines démontrent que les homologues des domaines insérés et des domaines parents peuvent exister indépendamment. Un exemple est celui des « doigts » insérés dans le domaine « paume » au sein des polymérases de la famille Pol I. Puisqu'un domaine peut être inséré dans un autre, il devrait toujours y avoir au moins un domaine continu dans une protéine multidomaine. C'est la principale différence entre les définitions des domaines structurels et des domaines évolutifs/fonctionnels. Un domaine évolutif sera limité à une ou deux connexions entre domaines, tandis que les domaines structurels peuvent avoir des connexions illimitées, dans le cadre d'un critère donné d'existence d'un noyau commun. Plusieurs domaines structurels pourraient être attribués à un domaine évolutif.
Un superdomaine est constitué de deux ou plusieurs domaines conservés d'origine nominalement indépendante, mais hérités ultérieurement comme une seule unité structurelle/fonctionnelle. Ce superdomaine combiné peut apparaître dans diverses protéines qui ne sont pas liées par la seule duplication génique. Un exemple de superdomaine est la paire protéine tyrosine phosphatase – domaine C2 dans PTEN , la tensine , l'auxiline et la protéine membranaire TPTE2. Ce superdomaine se trouve dans les protéines des animaux, des plantes et des champignons. Une caractéristique clé du superdomaine PTP-C2 est la conservation des résidus d'acides aminés dans l'interface du domaine.
Les domaines sont des unités de pliage autonomes
Pliant
Le repliement des protéines : le problème non résolu : Depuis les travaux fondateurs d' Anfinsen au début des années 1960, l'objectif de comprendre complètement le mécanisme par lequel un polypeptide se replie rapidement dans sa conformation native stable reste insaisissable. De nombreuses études expérimentales sur le repliement ont beaucoup contribué à notre compréhension, mais les principes qui régissent le repliement des protéines sont toujours basés sur ceux découverts dans les toutes premières études sur le repliement. Anfinsen a montré que l'état natif d'une protéine est thermodynamiquement stable, la conformation étant à un minimum global de son énergie libre.
Le repliement est une recherche dirigée de l'espace conformationnel permettant à la protéine de se replier sur une échelle de temps biologiquement réalisable. Le paradoxe de Levinthal stipule que si une protéine de taille moyenne devait échantillonner toutes les conformations possibles avant de trouver celle qui a la plus faible énergie, l'ensemble du processus prendrait des milliards d'années. Les protéines se replient généralement entre 0,1 et 1000 secondes. Par conséquent, le processus de repliement des protéines doit être dirigé d'une manière ou d'une autre par une voie de repliement spécifique. Les forces qui dirigent cette recherche sont susceptibles d'être une combinaison d'influences locales et globales dont les effets se font sentir à différentes étapes de la réaction.
Les progrès des études expérimentales et théoriques ont montré que le repliement peut être envisagé en termes de paysages énergétiques, où la cinétique de repliement est considérée comme une organisation progressive d'un ensemble de structures partiellement repliées à travers lesquelles une protéine passe sur son chemin vers la structure repliée. Cela a été décrit en termes d' entonnoir de repliement , dans lequel une protéine dépliée a un grand nombre d'états conformationnels disponibles et il y a moins d'états disponibles pour la protéine repliée. Un entonnoir implique que pour le repliement des protéines, il y a une diminution de l'énergie et une perte d'entropie avec l'augmentation de la formation de structures tertiaires. La rugosité locale de l'entonnoir reflète des pièges cinétiques, correspondant à l'accumulation d'intermédiaires mal repliés. Une chaîne de repliement progresse vers des énergies libres intra-chaîne plus faibles en augmentant sa compacité. Les options conformationnelles de la chaîne se rétrécissent de plus en plus vers une structure native.
Avantage des domaines dans le repliement des protéines
L'organisation des protéines de grande taille par domaines structuraux représente un avantage pour le repliement des protéines, chaque domaine étant capable de se replier individuellement, accélérant le processus de repliement et réduisant une combinaison potentiellement importante d'interactions entre résidus. De plus, compte tenu de la distribution aléatoire observée des résidus hydrophobes dans les protéines, la formation de domaines semble être la solution optimale pour qu'une protéine de grande taille puisse enterrer ses résidus hydrophobes tout en gardant les résidus hydrophiles à la surface.
Cependant, le rôle des interactions entre domaines dans le repliement des protéines et dans l'énergétique de stabilisation de la structure native diffère probablement pour chaque protéine. Dans le lysozyme T4, l'influence d'un domaine sur l'autre est si forte que la molécule entière est résistante au clivage protéolytique. Dans ce cas, le repliement est un processus séquentiel dans lequel le domaine C-terminal doit se replier indépendamment dans une étape précoce, et l'autre domaine nécessite la présence du domaine C-terminal replié pour le repliement et la stabilisation.
Il a été constaté que le repliement d'un domaine isolé peut se produire à la même vitesse ou parfois plus rapidement que celui du domaine intégré, suggérant que des interactions défavorables avec le reste de la protéine peuvent se produire pendant le repliement. Plusieurs arguments suggèrent que l'étape la plus lente du repliement des grandes protéines est l'appariement des domaines repliés. Cela est dû soit au fait que les domaines ne sont pas entièrement pliés correctement, soit au fait que les petits ajustements nécessaires à leur interaction sont défavorables sur le plan énergétique, comme l'élimination de l'eau de l'interface du domaine.
Domaines et flexibilité des protéines
La dynamique des domaines protéiques joue un rôle clé dans une multitude de processus de reconnaissance et de signalisation moléculaires. Les domaines protéiques, connectés par des domaines de liaison flexibles intrinsèquement désordonnés, induisent une allostérie à longue portée via la dynamique des domaines protéiques . Les modes dynamiques résultants ne peuvent généralement pas être prédits à partir des structures statiques de la protéine entière ou des domaines individuels. Ils peuvent cependant être déduits en comparant différentes structures d'une protéine (comme dans la base de données des mouvements moléculaires ). Ils peuvent également être suggérés par échantillonnage dans des trajectoires de dynamique moléculaire extensives et une analyse en composantes principales, ou ils peuvent être observés directement à l'aide de spectres mesurés par spectroscopie d'écho de spin neutronique .
Définition du domaine à partir des coordonnées structurelles
L'importance des domaines en tant que blocs de construction structuraux et éléments d'évolution a donné lieu à de nombreuses méthodes automatisées pour leur identification et leur classification dans les protéines de structure connue. Les procédures automatiques d'attribution fiable de domaines sont essentielles pour la génération de bases de données de domaines, en particulier à mesure que le nombre de structures protéiques connues augmente. Bien que les limites d'un domaine puissent être déterminées par inspection visuelle, la construction d'une méthode automatisée n'est pas simple. Des problèmes surviennent lorsqu'il s'agit de domaines discontinus ou fortement associés. Le fait qu'il n'existe pas de définition standard de ce qu'est réellement un domaine signifie que les attributions de domaines varient énormément, chaque chercheur utilisant un ensemble unique de critères.
Un domaine structural est une sous-structure compacte et globulaire comportant plus d'interactions en son sein qu'avec le reste de la protéine. Par conséquent, un domaine structural peut être déterminé par deux caractéristiques visuelles : sa compacité et son degré d'isolement. Des mesures de compacité locale dans les protéines ont été utilisées dans de nombreuses méthodes précoces d'attribution de domaines et dans plusieurs des méthodes les plus récentes.
Méthodes
L'un des premiers algorithmes utilisait une carte de distance Cα-Cα ainsi qu'une routine de clustering hiérarchique qui considérait les protéines comme plusieurs petits segments, d'une longueur de 10 résidus. Les segments initiaux étaient regroupés les uns après les autres en fonction des distances inter-segments ; les segments avec les distances les plus courtes étaient regroupés et considérés comme des segments uniques par la suite. Le clustering par étapes incluait finalement la protéine entière. Go a également exploité le fait que les distances inter-domaines sont normalement plus grandes que les distances intra-domaines ; toutes les distances Cα-Cα possibles étaient représentées sous forme de tracés diagonaux dans lesquels il y avait des modèles distincts pour les hélices, les brins étendus et les combinaisons de structures secondaires.
La méthode de Sowdhamini et Blundell regroupe les structures secondaires d'une protéine en fonction de leurs distances Cα-Cα et identifie les domaines à partir du motif de leurs dendrogrammes . Comme la procédure ne considère pas la protéine comme une chaîne continue d'acides aminés, il n'y a aucun problème à traiter les domaines discontinus. Des nœuds spécifiques dans ces dendrogrammes sont identifiés comme des groupes structurels tertiaires de la protéine, ceux-ci incluent à la fois des structures super-secondaires et des domaines. L'algorithme DOMAK est utilisé pour créer la base de données de domaines 3Dee. Il calcule une « valeur de division » à partir du nombre de chaque type de contact lorsque la protéine est divisée arbitrairement en deux parties. Cette valeur de division est importante lorsque les deux parties de la structure sont distinctes.
La méthode de Wodak et Janin était basée sur les surfaces d'interface calculées entre deux segments de chaîne clivés à plusieurs reprises à différentes positions de résidus. Les surfaces d'interface ont été calculées en comparant les surfaces des segments clivés à celles de la structure native. Les limites de domaine potentielles peuvent être identifiées à un site où la surface d'interface était minimale. D'autres méthodes ont utilisé des mesures d'accessibilité au solvant pour calculer la compacité.
L'algorithme PUU intègre un modèle harmonique utilisé pour approximer la dynamique inter-domaines. Le concept physique sous-jacent est que de nombreuses interactions rigides se produiront dans chaque domaine et que des interactions lâches se produiront entre les domaines. Cet algorithme est utilisé pour définir des domaines dans la base de données de domaines FSSP .
Swindells (1995) a développé une méthode, DETECTIVE, pour l'identification de domaines dans les structures protéiques basée sur l'idée que les domaines ont un intérieur hydrophobe. Des déficiences ont été constatées lorsque des noyaux hydrophobes de différents domaines continuent à travers la région d'interface.
RigidFinder est une nouvelle méthode d'identification de blocs rigides de protéines (domaines et boucles) de deux conformations différentes. Les blocs rigides sont définis comme des blocs dans lesquels toutes les distances entre résidus sont conservées à travers les conformations.
La méthode RIBFIND développée par Pandurangan et Topf identifie les corps rigides dans les structures protéiques en effectuant un regroupement spatial des éléments structurels secondaires dans les protéines. Les corps rigides RIBFIND ont été utilisés pour adapter de manière flexible les structures protéiques aux cartes de densité de cryomicroscopie électronique .
Une méthode générale pour identifier les domaines dynamiques , c'est-à-dire les régions protéiques qui se comportent approximativement comme des unités rigides au cours des fluctuations structurelles, a été introduite par Potestio et al. et, entre autres applications, a également été utilisée pour comparer la cohérence des subdivisions de domaines basées sur la dynamique avec celles basées sur la structure standard. La méthode, appelée PiSQRD, est disponible publiquement sous la forme d'un serveur Web. Ce dernier permet aux utilisateurs de subdiviser de manière optimale les protéines à chaîne unique ou multimériques en domaines quasi-rigides en fonction des modes collectifs de fluctuation du système. Par défaut, ces derniers sont calculés via un modèle de réseau élastique ; alternativement, des espaces dynamiques essentiels pré-calculés peuvent être téléchargés par l'utilisateur.
Exemples de domaines
- Répétitions Armadillo : nommées d'après la protéine Armadillo de type β-caténine de la mouche à fruits Drosophila melanogaster .
- Domaine basique de fermeture à glissière à leucine ( domaine bZIP ) : présent dans de nombreuses protéines eucaryotes se liant à l'ADN . Une partie du domaine contient une région qui assure les propriétés de liaison à l'ADN spécifiques à la séquence et la fermeture à glissière à leucine qui est nécessaire à la dimérisation de deux régions de liaison à l'ADN. La région de liaison à l'ADN comprend un certain nombre d'acides aminés basiques tels que l'arginine et la lysine .
- Répétitions de cadhérine : les cadhérines fonctionnent comme des protéines d'adhésion cellule-cellule dépendantes du Ca 2+ . Les domaines de cadhérine sont des régions extracellulaires qui assurent la liaison homophile de cellule à cellule entre les cadhérines à la surface des cellules adjacentes.
- Domaine effecteur de mort cellulaire (DED) : permet la liaison protéine-protéine par interactions homotypiques (DED-DED). Les protéases caspases déclenchent l'apoptose via des cascades protéolytiques. La pro-caspase-8 et la pro-caspase-9 se lient à des molécules adaptatrices spécifiques via les domaines DED, ce qui conduit à l'autoactivation des caspases.
- Main EF : un motif structurel hélice-tour-hélice trouvé dans chaque domaine structurel de la protéine de signalisation calmoduline et dans la protéine musculaire troponine-C .
- Domaine Foldon : un petit domaine protéique de la fibritine dans le bactériophage T4 qui peut provoquer la trimérisation des protéines.
- Domaines de type immunoglobuline : présents dans les protéines de la superfamille des immunoglobulines (IgSF). Ils contiennent environ 70 à 110 acides aminés et sont classés en différentes catégories (IgV, IgC1, IgC2 et IgI) selon leur taille et leur fonction. Ils possèdent un pli caractéristique dans lequel deux feuillets bêta forment un « sandwich » stabilisé par des interactions entre des cystéines conservées et d'autres acides aminés chargés . Ils sont importants pour les interactions protéine-protéine dans les processus d' adhésion cellulaire , d'activation cellulaire et de reconnaissance moléculaire. Ces domaines se trouvent généralement dans les molécules jouant un rôle dans le système immunitaire .
- Domaine de liaison à la phosphotyrosine (PTB) : les domaines PTB se lient généralement aux résidus de tyrosine phosphorylés. On les trouve souvent dans les protéines de transduction du signal. La spécificité de liaison du domaine PTB est déterminée par les résidus du côté amino-terminal de la phosphotyrosine. Exemples : les domaines PTB de SHC et d'IRS-1 se lient à une séquence NPXpY . Les protéines contenant du PTB telles que SHC et IRS-1 sont importantes pour les réponses à l'insuline des cellules humaines.
- Domaine d'homologie de la pleckstrine (PH) : les domaines PH se lient aux phosphoinositides avec une affinité élevée. Une spécificité pour PtdIns(3)P , PtdIns(4)P, PtdIns(3,4)P2 , PtdIns(4,5)P2 et PtdIns(3,4,5)P3 a été observée. Étant donné que les phosphoinositides sont séquestrés dans diverses membranes cellulaires (en raison de leur longue queue lipophile), les domaines PH provoquent généralement le recrutement de la protéine en question vers une membrane où la protéine peut exercer une certaine fonction dans la signalisation cellulaire, la réorganisation du cytosquelette ou le trafic membranaire.
- Domaine d'homologie 2 de Src (SH2) : les domaines SH2 sont souvent présents dans les protéines de transduction du signal. Les domaines SH2 confèrent une liaison à la tyrosine phosphorylée (pTyr). Nommé d'après le domaine de liaison à la phosphotyrosine de l' oncogène viral src , qui est lui-même une tyrosine kinase . Voir également : domaine SH3 .
- Domaine de liaison à l'ADN du doigt de zinc (ZnF_GATA) : les protéines contenant le domaine ZnF_GATA sont généralement des facteurs de transcription qui se lient généralement à la séquence d'ADN [AT]GATA[AG] des promoteurs .
Domaines de fonction inconnue
Une grande partie des domaines ont une fonction inconnue. Un domaine de fonction inconnue (DUF) est un domaine protéique qui n'a pas de fonction caractérisée. Ces familles ont été rassemblées dans la base de données Pfam à l'aide du préfixe DUF suivi d'un numéro, les exemples étant DUF2992 et DUF1220. Il existe désormais plus de 3 000 familles DUF dans la base de données Pfam, ce qui représente plus de 20 % des familles connues. Étonnamment, le nombre de DUF dans Pfam est passé de 20 % (en 2010) à 22 % (en 2019), principalement en raison d'un nombre croissant de nouvelles séquences génomiques . La version 32.0 de Pfam (2019) contenait 3 961 DUF.