Le facteur de croissance endothélial vasculaire ( VEGF , / v ɛ dʒ ˈ ɛ f / ), connu à l'origine sous le nom de facteur de perméabilité vasculaire ( VPF ), est une protéine de signalisation produite par de nombreuses cellules qui stimule la formation de vaisseaux sanguins. Pour être précis, le VEGF est une sous-famille de facteurs de croissance , la famille des facteurs de croissance dérivés des plaquettes des facteurs de croissance à nœuds cystéiniques . Ce sont des protéines de signalisation importantes impliquées à la fois dans la vasculogenèse (la formation de novo du système circulatoire embryonnaire ) et dans l'angiogenèse (la croissance des vaisseaux sanguins à partir d'un système vasculaire préexistant).
Il fait partie du système qui rétablit l'apport d'oxygène aux tissus lorsque la circulation sanguine est insuffisante, comme dans des conditions hypoxiques. La concentration sérique de VEGF est élevée dans l'asthme bronchique et le diabète sucré . La fonction normale du VEGF est de créer de nouveaux vaisseaux sanguins pendant le développement embryonnaire , de nouveaux vaisseaux sanguins après une blessure, des muscles après un exercice et de nouveaux vaisseaux ( circulation collatérale ) pour contourner les vaisseaux bloqués. Il peut contribuer à la maladie. Les cancers solides ne peuvent pas se développer au-delà d'une taille limitée sans un apport sanguin adéquat ; les cancers qui peuvent exprimer le VEGF sont capables de se développer et de métastaser. La surexpression du VEGF peut provoquer une maladie vasculaire de la rétine de l'œil et d'autres parties du corps. Des médicaments tels que l'aflibercept , le bévacizumab , le ranibizumab et le pegaptanib peuvent inhiber le VEGF et contrôler ou ralentir ces maladies.
Histoire
En 1970, Judah Folkman et al . ont décrit un facteur sécrété par les tumeurs provoquant l'angiogenèse et l'ont appelé facteur d'angiogenèse tumorale . En 1983, Senger et al. ont identifié un facteur de perméabilité vasculaire sécrété par les tumeurs chez les cobayes et les hamsters. En 1989, Ferrara et Henzel ont décrit un facteur identique dans les cellules folliculaires hypophysaires bovines qu'ils ont purifiées, clonées et nommées VEGF. Un épissage alternatif similaire du VEGF a été découvert par Tischer et al. en 1991. Entre 1996 et 1997, Christinger et De Vos ont obtenu la structure cristalline du VEGF, d'abord à une résolution de 2,5 Å, puis à 1,9 Å.
En 1992, Ferrara et al. ont montré que la tyrosine kinase-1 (flt-1) de type Fms était un récepteur du VEGF. et al. ont également montré que le récepteur du domaine d'insertion de la kinase (KDR) était un récepteur du VEGF . En 1998, il a été montré que la neuropiline 1 et la neuropiline 2 agissaient comme des récepteurs du VEGF.
Classification
Chez les mammifères, la famille VEGF comprend cinq membres : VEGF-A , le facteur de croissance placentaire ( PGF ), VEGF-B , VEGF-C et VEGF-D . Ces derniers membres ont été découverts après VEGF-A ; avant leur découverte, VEGF-A était connu sous le nom de VEGF. Un certain nombre de protéines apparentées au VEGF codées par des virus (VEGF-E) et dans le venin de certains serpents (VEGF-F) ont également été découvertes.
L'activité du VEGF-A, comme son nom l'indique, a été étudiée principalement sur les cellules de l' endothélium vasculaire , bien qu'il ait des effets sur un certain nombre d'autres types de cellules (par exemple, stimulation de la migration des monocytes / macrophages , neurones, cellules cancéreuses, cellules épithéliales rénales). In vitro, il a été démontré que le VEGF-A stimule la mitogenèse des cellules endothéliales et la migration cellulaire . Le VEGF-A est également un vasodilatateur et augmente la perméabilité microvasculaire et était à l'origine appelé facteur de perméabilité vasculaire.
Isoformes
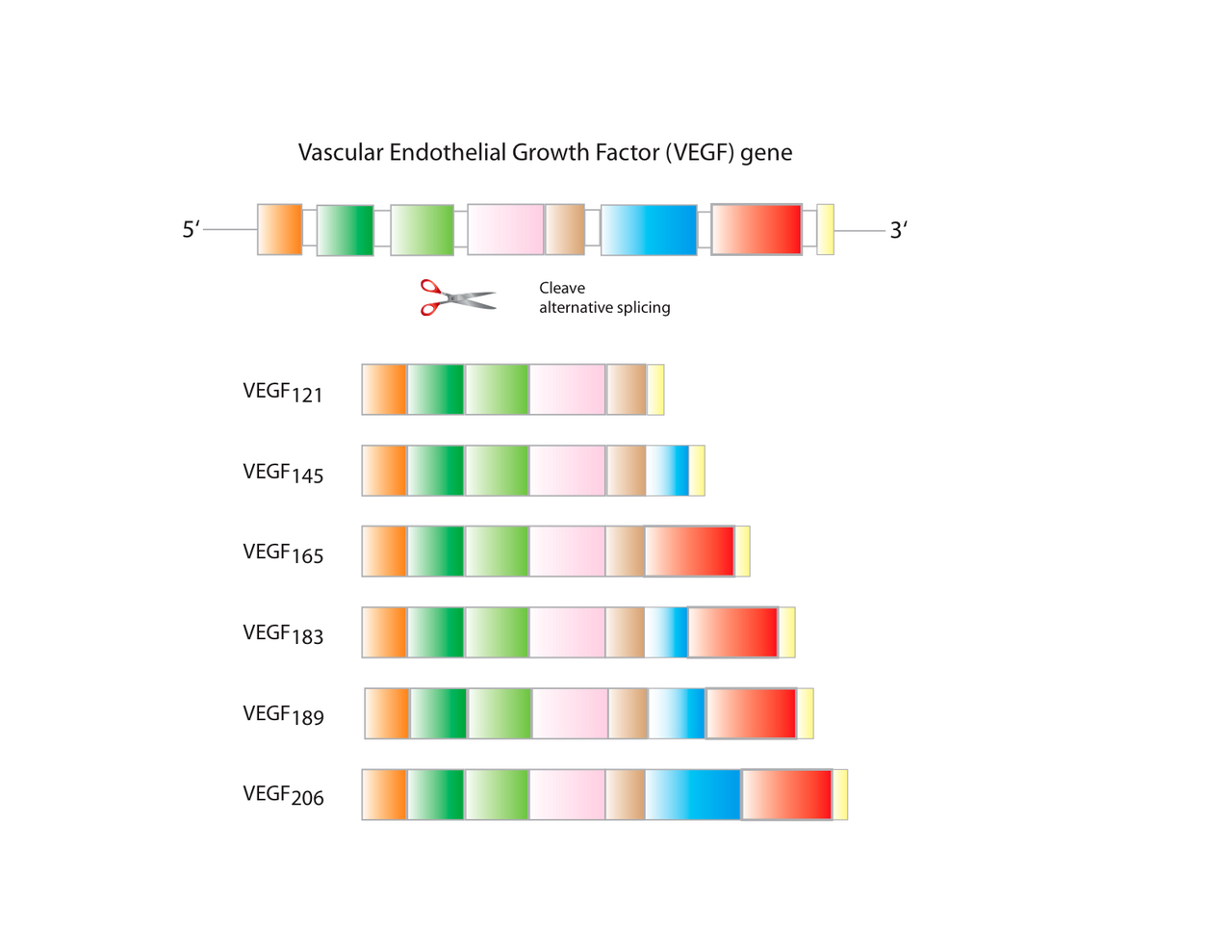
Il existe plusieurs isoformes de VEGF-A qui résultent de l'épissage alternatif de l'ARNm d'un seul gène VEGFA à 8 exons . Ceux-ci sont classés en deux groupes qui sont désignés en fonction de leur site d'épissage de l'exon terminal (exon 8) : le site d'épissage proximal (noté VEGF xxx ) ou le site d'épissage distal (VEGF xxx b). De plus, l'épissage alterné des exons 6 et 7 modifie leur affinité de liaison à l'héparine et leur nombre d'acides aminés (chez l'homme : VEGF 121 , VEGF 121 b, VEGF 145 , VEGF 165 , VEGF 165 b, VEGF 189 , VEGF 206 ; les orthologues de ces protéines chez les rongeurs contiennent un acide aminé de moins). Ces domaines ont des conséquences fonctionnelles importantes pour les variantes d'épissage du VEGF, car le site d'épissage terminal (exon 8) détermine si les protéines sont pro-angiogéniques (site d'épissage proximal, exprimé pendant l'angiogenèse) ou anti-angiogéniques (site d'épissage distal, exprimé dans les tissus normaux). De plus, l'inclusion ou l'exclusion des exons 6 et 7 favorisent les interactions avec les protéoglycanes à sulfate d'héparane (HSPG) et les co-récepteurs de neuropiline à la surface cellulaire, améliorant leur capacité à se lier et à activer les récepteurs du VEGF (VEGFR). Récemment, il a été démontré que le VEGF-C est un inducteur important de la neurogenèse dans la zone sous-ventriculaire murine, sans exercer d'effets angiogéniques.
Mécanisme
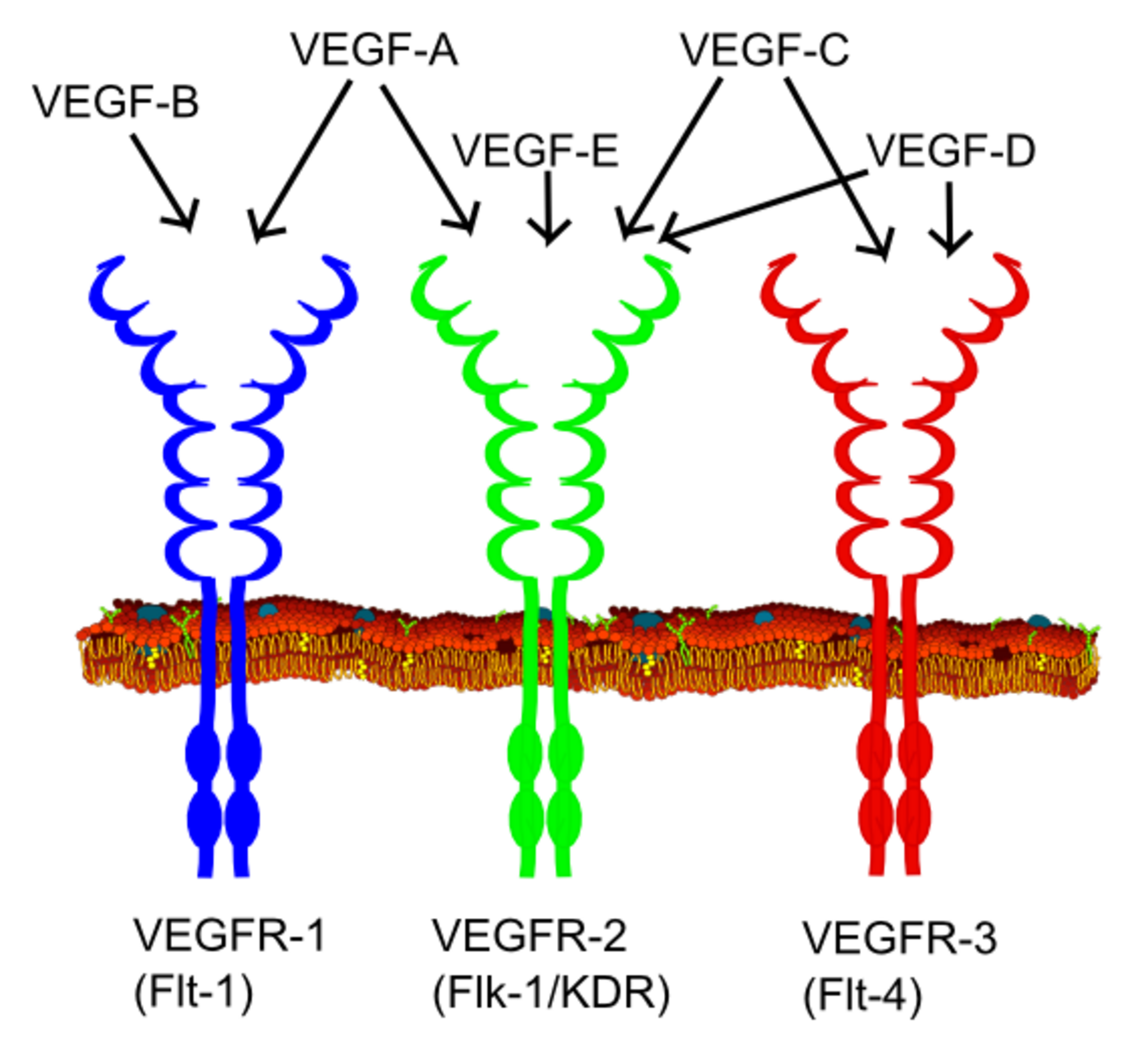
Tous les membres de la famille VEGF stimulent les réponses cellulaires en se liant aux récepteurs de tyrosine kinase (les VEGFR ) à la surface cellulaire, ce qui les amène à se dimériser et à s'activer par transphosphorylation , bien qu'à des sites, des moments et des degrés différents. Les récepteurs VEGF ont une partie extracellulaire composée de 7 domaines de type immunoglobuline, une seule région transmembranaire et une partie intracellulaire contenant un domaine tyrosine kinase divisé. Le VEGF-A se lie au VEGFR-1 ( Flt-1 ) et au VEGFR-2 ( KDR/Flk-1 ). Le VEGFR-2 semble être le médiateur de presque toutes les réponses cellulaires connues au VEGF. La fonction du VEGFR-1 est moins bien définie, bien qu'on pense qu'il module la signalisation du VEGFR-2. Une autre fonction du VEGFR-1 peut être d'agir comme un récepteur factice/leurre, séquestrant le VEGF de la liaison au VEGFR-2 (cela semble être particulièrement important pendant la vasculogenèse dans l'embryon). Le VEGF-C et le VEGF-D, mais pas le VEGF-A, sont des ligands d'un troisième récepteur ( VEGFR-3/Flt4 ), qui assure la médiation de la lymphangiogenèse . Le récepteur (VEGFR3) est le site de liaison des ligands principaux (VEGFC et VEGFD), qui assure la médiation de l'action perpétuelle et de la fonction des ligands sur les cellules cibles. Le facteur de croissance endothélial vasculaire-C peut stimuler la lymphangiogenèse (via VEGFR3) et l'angiogenèse via VEGFR2. Le facteur de croissance endothélial vasculaire-R3 a été détecté dans les cellules endothéliales lymphatiques dans la lymphangiogenèse de nombreuses espèces, bovins, buffles et primates.
En plus de se lier aux VEGFR , le VEGF se lie à des complexes récepteurs constitués à la fois de neuropilines et de VEGFR. Ce complexe récepteur a augmenté l'activité de signalisation du VEGF dans les cellules endothéliales ( vaisseaux sanguins ). Les neuropilines (NRP) sont des récepteurs pléiotropes et par conséquent d'autres molécules peuvent interférer avec la signalisation des complexes récepteurs NRP/VEGFR. Par exemple, les sémaphorines de classe 3 entrent en compétition avec le VEGF 165 pour la liaison au NRP et pourraient donc réguler l'angiogenèse médiée par le VEGF .
Expression
La production de VEGF-A peut être induite dans une cellule qui ne reçoit pas suffisamment d'oxygène . Lorsqu'une cellule manque d'oxygène, elle produit du HIF, un facteur inductible par l'hypoxie , un facteur de transcription. Le HIF stimule la libération de VEGF-A, entre autres fonctions (y compris la modulation de l'érythropoïèse). Le VEGF-A circulant se lie ensuite aux récepteurs du VEGF sur les cellules endothéliales, déclenchant une voie de tyrosine kinase conduisant à l'angiogenèse. L'expression de l'angiopoïétine-2 en l'absence de VEGF conduit à la mort des cellules endothéliales et à la régression vasculaire. À l'inverse, une étude allemande réalisée in vivo a révélé que les concentrations de VEGF diminuaient en fait après une réduction de 25 % de l'apport en oxygène pendant 30 minutes. HIF1 alpha et HIF1 bêta sont constamment produits, mais HIF1 alpha est très labile en O 2 , donc, dans des conditions aérobies, il est dégradé. Lorsque la cellule devient hypoxique, HIF1 alpha persiste et le complexe HIF1alpha/beta stimule la libération de VEGF. L'utilisation combinée de microvésicules et de 5-FU a entraîné une chimiosensibilité accrue des cellules du carcinome épidermoïde plus que l'utilisation de 5-FU ou de microvésicules seules. De plus, la régulation négative de l'expression du gène VEGF a été associée à une diminution de l'expression du gène CD1.
Importance clinique
En cas de maladie
Le VEGF-A et les récepteurs correspondants sont rapidement régulés à la hausse après une lésion traumatique du système nerveux central (SNC). Le VEGF-A est fortement exprimé dans les stades aigus et subaigus de la lésion du SNC, mais l'expression de la protéine diminue avec le temps. Cette durée d'expression du VEGF-A correspond à la capacité de revascularisation endogène après la lésion. Cela suggère que le VEGF-A / VEGF 165 pourrait être utilisé comme cible pour favoriser l'angiogenèse après des lésions traumatiques du SNC. Cependant, il existe des rapports scientifiques contradictoires sur les effets des traitements par VEGF-A dans les modèles de lésions du SNC.
Bien qu'il n'ait pas été associé comme biomarqueur pour le diagnostic d' accident vasculaire cérébral ischémique aigu , des niveaux élevés de VEGF sérique dans les 48 premières heures après un infarctus cérébral ont été associés à un mauvais pronostic après 6 mois et 2 ans.
Le VEGF-A a été associé à un mauvais pronostic dans le cancer du sein . De nombreuses études montrent une diminution de la survie globale et de la survie sans maladie dans les tumeurs surexprimant le VEGF. La surexpression du VEGF-A peut être une étape précoce du processus de métastase , une étape impliquée dans le changement « angiogénique ». Bien que le VEGF-A ait été corrélé à une faible survie, son mécanisme d'action exact dans la progression des tumeurs reste flou.
Le VEGF-A est également libéré dans la polyarthrite rhumatoïde en réponse au TNF-α , augmentant la perméabilité endothéliale et le gonflement et stimulant également l'angiogenèse (formation de capillaires).
Le VEGF-A est également important dans la rétinopathie diabétique (RD). Les problèmes de microcirculation dans la rétine des personnes atteintes de diabète peuvent provoquer une ischémie rétinienne, qui entraîne la libération de VEGF-A et un changement d'équilibre des isoformes pro-angiogéniques VEGF xxx par rapport aux isoformes VEGF xxx b normalement exprimées. Le VEGF xxx peut alors provoquer la création de nouveaux vaisseaux sanguins dans la rétine et ailleurs dans l'œil, annonçant des changements qui peuvent menacer la vue.
Le VEGF-A joue un rôle dans la pathologie de la forme humide de la dégénérescence maculaire liée à l'âge (DMLA), principale cause de cécité chez les personnes âgées dans les pays industrialisés. La pathologie vasculaire de la DMLA présente certaines similitudes avec la rétinopathie diabétique, bien que la cause de la maladie et la source typique de néovascularisation diffèrent entre les deux maladies.
Les taux sériques de VEGF-D sont significativement élevés chez les patients atteints d'angiosarcome .
Une fois libéré, le VEGF-A peut déclencher plusieurs réponses. Il peut permettre à une cellule de survivre, de se déplacer ou de se différencier davantage. Le VEGF est donc une cible potentielle pour le traitement du cancer . Le premier médicament anti-VEGF, un anticorps monoclonal appelé bevacizumab , a été approuvé en 2004. Environ 10 à 15 % des patients bénéficient d'un traitement par bevacizumab ; cependant, les biomarqueurs de l'efficacité du bevacizumab ne sont pas encore connus.
Des études récentes montrent que les VEGF ne sont pas les seuls promoteurs de l'angiogenèse. En particulier, le FGF2 et le HGF sont de puissants facteurs angiogéniques.
On a constaté que les patients souffrant d’emphysème pulmonaire présentaient des niveaux réduits de VEGF dans les artères pulmonaires.
Il a également été démontré que le VEGF-D est surexprimé dans la lymphangioléiomyomatose et est actuellement utilisé comme biomarqueur diagnostique dans le traitement de cette maladie rare.
Dans le rein , l’expression accrue du VEGF-A dans les glomérules provoque directement l’hypertrophie glomérulaire associée à la protéinurie.
Les altérations du VEGF peuvent être prédictives d’ une prééclampsie à début précoce .
Les thérapies géniques pour l’angine réfractaire établissent l’expression du VEGF dans les cellules épicardiques pour favoriser l’angiogenèse.