Les leucodystrophies sont un groupe de troubles, généralement héréditaires, caractérisés par une dégénérescence de la substance blanche du cerveau. Le mot leucodystrophie vient des racines grecques leuko , « blanc », dys , « anormal » et troph , « croissance ». Les leucodystrophies sont causées par une croissance ou un développement imparfait des cellules gliales qui produisent la gaine de myéline , la gaine isolante graisseuse qui entoure les fibres nerveuses . Les leucodystrophies peuvent être classées comme maladies hypomyélinisantes ou démyélinisantes , respectivement, selon que les lésions sont présentes avant la naissance ou surviennent après. Alors que toutes les leucodystrophies sont le résultat de mutations génétiques, d'autres troubles démyélinisants ont une étiologie auto-immune , infectieuse ou métabolique .
Lorsque la substance blanche est endommagée, les réponses immunitaires qui s’ensuivent peuvent entraîner une inflammation du système nerveux central (SNC), ainsi qu’une perte de myéline. La dégénérescence de la substance blanche peut être observée à l’ IRM et est utilisée pour diagnostiquer la leucodystrophie. La leucodystrophie se caractérise par des symptômes spécifiques, notamment une diminution de la fonction motrice, une rigidité musculaire et une dégénérescence éventuelle de la vue et de l’ouïe. Bien que la maladie soit mortelle, l’âge d’apparition est un facteur clé, car les nourrissons ont une espérance de vie typique de 2 à 8 ans, tandis que les adultes vivent généralement plus d’une décennie après le début de la maladie. Les options de traitement sont limitées, bien que les transplantations de cellules souches hématopoïétiques à partir de moelle osseuse ou de sang de cordon ombilical semblent aider dans certains types de leucodystrophie, tandis que des recherches supplémentaires sont en cours.
L'incidence combinée des leucodystrophies est estimée à 1 sur 7 600. La majorité des types impliquent l'hérédité d'un trait récessif lié au chromosome X ou d'un trait dominant lié au chromosome X , tandis que d'autres, bien qu'impliquant un gène défectueux, sont le résultat d' une mutation spontanée plutôt que d'une hérédité génétique .
Symptômes et signes
Certains symptômes spécifiques varient d'un type de leucodystrophie à l'autre, mais la grande majorité des symptômes sont communs car les causes de la maladie ont généralement les mêmes effets. Les symptômes dépendent de l'âge d'apparition, qui se situe principalement dans la petite enfance et la petite enfance, bien que le moment exact d'apparition puisse être difficile à déterminer. L'hyperirritabilité et l'hypersensibilité à l'environnement sont courantes, ainsi que certains signes physiques révélateurs, notamment une rigidité musculaire et une tête penchée vers l'arrière. La thérapie au Botox est souvent utilisée pour traiter les patients atteints de spasticité. Les débuts juvéniles et adultes présentent des symptômes similaires, notamment une diminution ou une perte de l'audition et de la vision. Bien que les enfants souffrent de dégénérescence optique et auditive, l'évolution de la maladie est généralement trop rapide, entraînant la mort relativement rapidement, alors que les adultes peuvent vivre avec ces conditions pendant de nombreuses années. Chez les enfants, l'activité spastique précède souvent une ataxie progressive et une détérioration cognitive rapide qui a été décrite comme un retard mental . L'épilepsie est courante chez les patients de tous âges. Les patients plus avancés présentent une faiblesse de la déglutition , ce qui entraîne des quintes de toux spasmodique dues à l'inhalation de salive. La progression symptomatique classique de l'adrénoleucodystrophie juvénile liée à l'X est illustrée dans le film de 1992, Lorenzo's Oil .
L'évolution et le calendrier dépendent de l'âge d'apparition, les nourrissons ayant une espérance de vie de 2 à 8 ans, les adolescents de 2 à 10 ans et les adultes généralement de 10 ans et plus. Les adultes connaissent généralement une période prolongée de stabilité suivie d'un déclin vers un état végétatif et la mort. Bien que des traitements existent, la plupart sont en phase expérimentale et ne peuvent que promettre un arrêt de la progression des symptômes, bien que certaines thérapies géniques aient montré une certaine amélioration symptomatique. L'évolution débilitante de la maladie a donné lieu à de nombreux débats philosophiques et éthiques sur les essais cliniques expérimentaux, les droits des patients et le suicide assisté par un médecin .
Causes
Bien que les causes sous-jacentes les plus spécifiques de la leucodystrophie dépendent du type, il existe des schémas physiopathologiques communs qui peuvent être observés parmi tous les types. Tout d'abord, la leucodystrophie est une maladie neurodégénérative qui résulte toujours à la fois de la détérioration et de l'entretien des gaines de myéline entourant les axones neuronaux dans le système nerveux central à la suite d'une mutation génétique . La myéline est une substance blanche grasse qui agit comme un isolant électrique et recouvre les axones afin d'accélérer les impulsions (c'est-à-dire les potentiels d'action ) qui se déplacent le long de l'axone. Ainsi, le résultat naturel d'une perte de cette substance est une diminution de l'efficacité de la propagation des impulsions. Comme la myéline est produite par les oligodendrocytes (un type de cellule gliale ) dans le système nerveux central, un endroit facile à rechercher pour la cause est une mutation ou un dysfonctionnement de ces cellules et d'autres cellules gliales.
Influence génétique
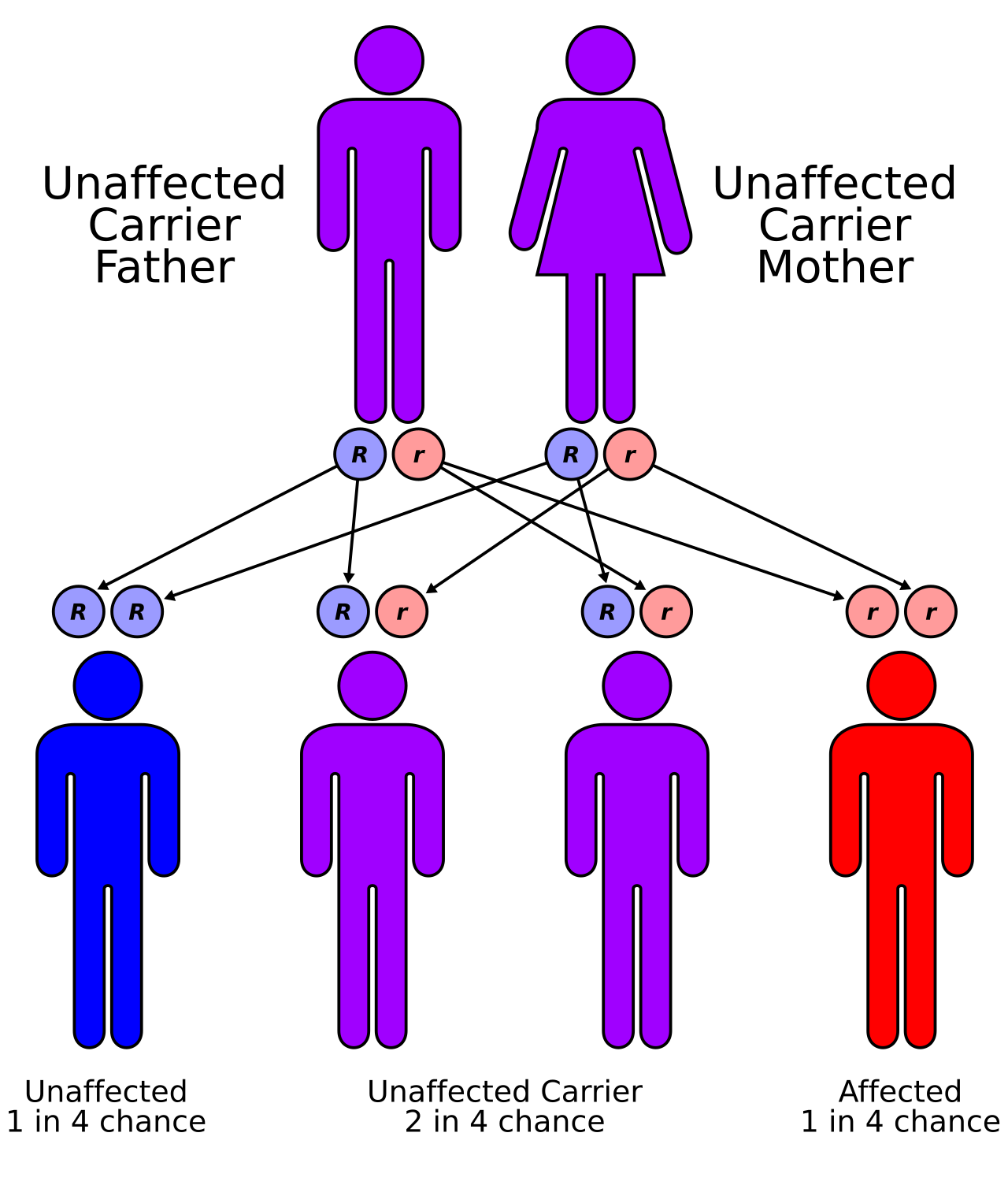
Les formes héréditaires de leucodystrophie sont généralement le résultat d'un modèle de transmission autosomique récessif , bien que les modèles de transmission dominants ne soient pas rares, comme dans le cas de la leucodystrophie à début adulte. Cela signifie que l' allèle affecté est porté sur un chromosome autosomique , ou non sexuel, et est masqué par le phénotype dominant non affecté . En d'autres termes, pour qu'un individu hérite du phénotype de leucodystrophie, il ou elle doit porter deux des allèles mutants récessifs. La maladie de Krabbe et la leucodystrophie métachromatique (MLD) sont deux de ce type. La MLD se trouve sur le chromosome humain 22 à la position q13.31. Un autre type de leucodystrophie héréditaire est l'adrénoleucodystrophie liée à l'X (X-ALD). Comme son nom l'indique, ce type de leucodystrophie est le résultat d'une mutation trouvée sur le chromosome X. Elle est également portée selon un modèle récessif. Le chromosome X est un chromosome sexuel et, comme les femmes ont deux « chances » d'acquérir un chromosome X normal (un chromosome maternel, un chromosome paternel) et les hommes une seule chance (un chromosome maternel), cette maladie est plus susceptible d'être observée chez les hommes que chez les femmes. La mutation entraînant la leucodystrophie à l'âge adulte est localisée au niveau du chromosome 5q23.
Physiopathologie
Bien qu'il existe près de 40 types différents de leucodystrophie, beaucoup d'entre eux ne font pas l'objet de recherches formelles et approfondies. La plupart des recherches menées jusqu'à présent ont porté sur cinq types : (1) la leucodystrophie métachromatique (MLD), (2) la maladie de Krabbe , (3) l'adrénoleucodystrophie liée à l'X (ALD), (4) la maladie de Canavan et (5) la maladie d'Alexander . Chaque type de leucodystrophie a une physiopathologie unique , mais tous ces cinq types affectent d'une manière ou d'une autre un sous-ensemble de cellules gliales, perturbant ainsi la production et le maintien de la myéline, et impliquent généralement une mutation impliquant des gènes codant pour des enzymes nécessaires au catabolisme des acides gras à très longue chaîne (VLCFA) qui sont toxiques pour les cellules productrices de myéline du système nerveux central.
Leucodystrophie métachromatique
La leucodystrophie métachromatique est le résultat de défauts génétiques dans les enzymes associées au compartiment cellulaire appelé lysosome . La MLD est l'une des deux leucodystrophies qui sont également un trouble de stockage lysosomal . La MLD est héritée de manière autosomique récessive et résulte de mutations dans trois allèles ARSA différents qui codent l'enzyme arylsulfatase A (ASA ou parfois ARSA), également appelée sulfatide sulfatase . L'AAS est responsable de la dégradation des sulfatides, des sphingolipides présents dans les membranes neuronales ainsi que dans la myéline. Lorsqu'il y a une mutation dans le gène qui code l'AAS, elle diminue la production d'AAS, ce qui entraîne par la suite une diminution de la dégradation des sulfatides, provoquant ainsi leur accumulation. Cette accumulation de sulfatides est toxique pour les oligodendrocytes, les cellules productrices de myéline du SNC, ce qui entraîne une perturbation de la structure de la myéline suivie d' une démyélinisation . Le mode d'hérédité des trois différents allèles affecte le type de MLD qu'une personne développe. Deux allèles nuls sont responsables de la version infantile et ne permettent aucune production d'AAS. Un individu hétérozygote (un allèle nul, un allèle non nul) développe la forme juvénile et a une certaine production d'AAS, tandis qu'un individu avec deux allèles non nuls mutés développe la forme adulte.
Maladie de Krabbe
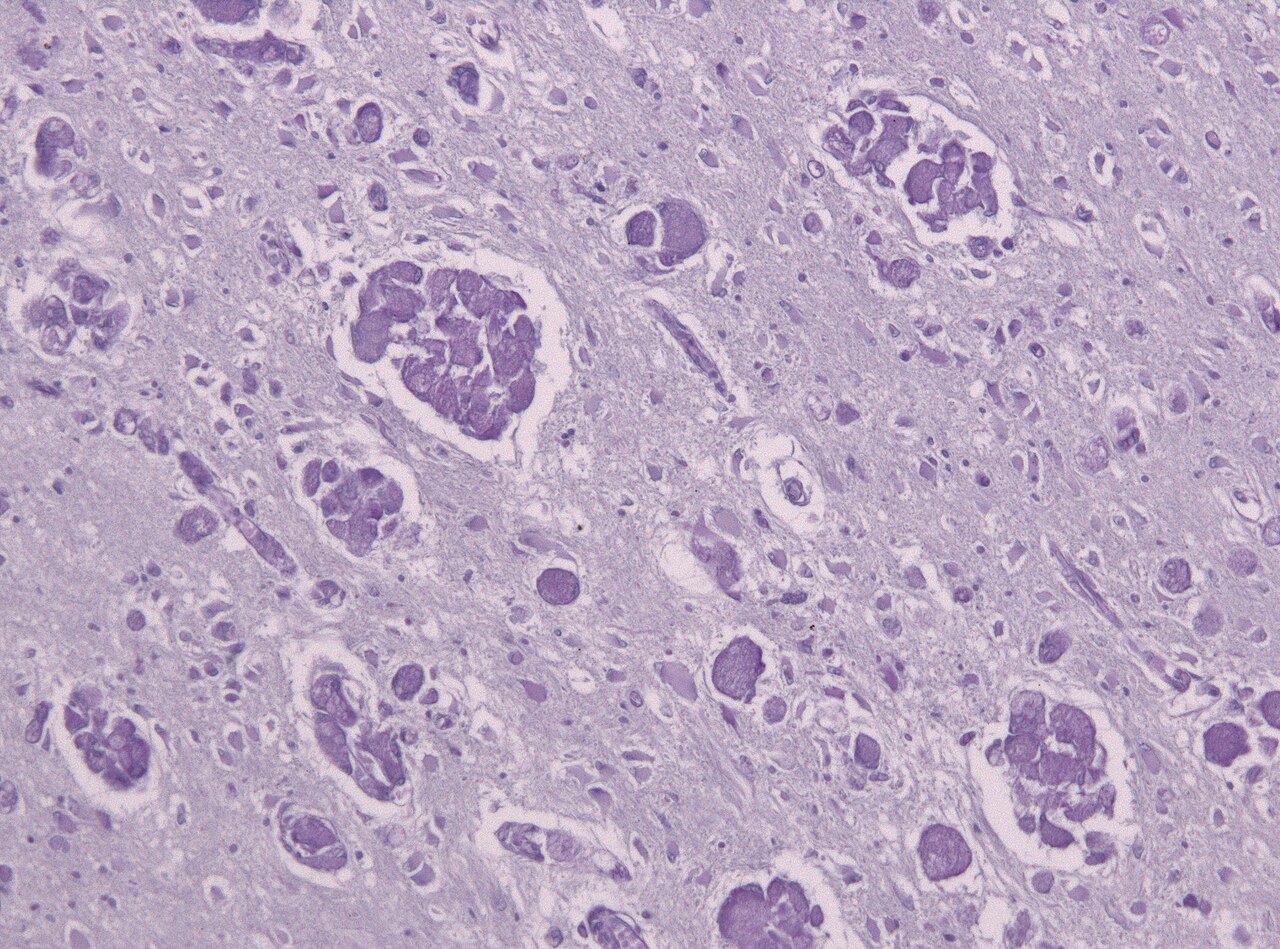
Comme la MLD, la maladie de Krabbe est un autre type de leucodystrophie à transmission autosomique récessive qui résulte d'un trouble de surcharge lysosomale . Elle est due à une délétion dans l'exon 16 du gène GALC qui provoque une mutation par décalage du cadre de lecture conduisant à un codon stop prématuré . Le gène GALC, situé sur le chromosome 14 en position 31 (14q31), code pour l' enzyme bêta-galactocérébrosidase (GALC). La GALC est une enzyme lysosomale responsable du catabolisme des galactolipides , en particulier du lipide toxique psychosine , qui sont largement distribués dans tout le cerveau. Une déficience en GALC provoque ainsi une accumulation de ces acides gras , conduisant à une incursion de cellules appelées « macrophages globoïdes » qui détruisent les oligodendrocytes, inhibant ainsi toute formation supplémentaire de myéline. Étant donné la présence de macrophages globoïdes regroupés près de la matière blanche , la maladie de Krabbe est souvent appelée leucodystrophie à cellules globoïdes.
Maladie de Canavan
La maladie de Canavan est un type de leucodystrophie moins étudié qui, comme la MLD et la maladie de Krabbe, est également héritée selon un modèle autosomique récessif. Elle est due à une mutation du gène ASPA qui code l'aspartoacylase , une enzyme nécessaire au métabolisme du N-acétyl-L-aspartate (NAA). La mutation provoque un déficit en aspartoacyclase. La NAA est impliquée dans la formation des lipides ; si elle n'est pas dégradée par l'aspartoacylase, les taux de lipides dans le cerveau augmentent, provoquant une démyélinisation.
Adrénoleucodystrophie liée à l'X
Dans l'adrénoleucodystrophie liée au chromosome X (X-ALD), une mutation se produit dans la cassette de liaison de l'ATP peroxysomale ( transporteur ABC ). Cela conduit à une démyélinisation inflammatoire cérébrale causée par une déstabilisation de la myéline. La démyélinisation inflammatoire commence dans le corps calleux et progresse lentement vers l'extérieur dans les deux hémisphères. Chez les patients atteints de X-ALD, des niveaux anormalement élevés de VLCFA s'accumulent dans divers tissus et fluides corporels. Cette concentration accrue s'intègre ensuite dans divers lipides complexes où les VLCFA ne se trouvent normalement pas. Il a été découvert que cela est directement impliqué dans l'inflammation cérébrale de la X-ALD. On suppose que les VLCFA accumulés et intégrés dans les lipides complexes pourraient conduire à la déstabilisation de la gaine de myéline et éventuellement à la démyélinisation.
La maladie d'Alexandre
La maladie d'Alexander est différente des leucodystrophies mentionnées ci-dessus, car elle résulte d' une mutation spontanée , ce qui signifie qu'elle n'est pas héréditaire. La mutation trouvée chez un individu affecté ne se retrouve pas chez aucun de ses parents. Les symptômes résultent de l'accumulation de protéine acide fibrillaire gliale (GFAP) à la suite d'une mutation du gène GFAP , dont la protéine, plutôt que d'être trouvée en association avec les lysosomes ou les peroxysomes, est un filament intermédiaire lié à l' enveloppe nucléaire . Les filaments intermédiaires sont des protéines responsables de la constitution du cytosquelette cellulaire ; ainsi, ce type de mutation provoque un développement structurel anormal des cellules d'une personne. Des défauts du cytosquelette et des molécules de transport ont été observés dans les astrocytes des individus affectés. Ces astrocytes contiennent des niveaux anormalement élevés de protéine GFAP, affectant leur développement et leur fonction.
Diagnostic
La dégénérescence de la substance blanche , qui reflète la dégénérescence de la myéline, peut être observée dans une IRM de base et utilisée pour diagnostiquer les leucodystrophies de tous types. Les images FLAIR ( inversion recovery attenuated fluid-attenuated ) pondérées en T-1 et T-2 sont l'approche la plus souvent utilisée. Des tests électrophysiologiques et d'autres types de tests de laboratoire peuvent également être effectués. En particulier, la vitesse de conduction nerveuse est examinée pour faire la distinction entre la leucodystrophie et d'autres maladies démyélinisantes , ainsi que pour faire la distinction entre les leucodystrophies individuelles. Par exemple, les personnes atteintes de X-ALD ont des vitesses de conduction normales, tandis que celles atteintes de la maladie de Krabbe ou de leucodystrophie métachromatique ont des anomalies dans leurs vitesses de conduction. Des panels de séquençage multigénique pour la leucodystrophie indifférenciée sont proposés pour un diagnostic moléculaire rapide après un conseil génétique.
Types
Les types spécifiques de leucodystrophie comprennent les suivants avec leurs codes CIM-10 respectifs lorsqu'ils sont disponibles :
- (E75.2) Maladie d'Alexandre
- (E75.2) Maladie de Canavan
- (E75.2) Leucodystrophie hypomyélinisante de type 7 (syndrome 4H)
- (E75.2) Maladie de Krabbe
- (E75.2) Leucodystrophie métachromatique
- (E75.2) Maladie de Pelizaeus-Merzbacher
- (E75.5) Xanthomatose cérébrotendineuse
Traitement
Les différents types de leucodystrophies et leurs causes sont multiples et les traitements varient en fonction de chaque type. Des études et des essais cliniques tentent de trouver des traitements pour chacune des différentes leucodystrophies. Les greffes de cellules souches et la thérapie génique semblent être les plus prometteuses pour traiter toutes les leucodystrophies, à condition qu'elles soient pratiquées le plus tôt possible, avant l'apparition de lésions neurologiques importantes.
Pour les leucodystrophies hypomyélinisantes, la recherche thérapeutique sur les thérapies cellulaires semble prometteuse. Des cellules précurseurs d'oligodendrocytes et des cellules souches neurales ont été transplantées avec succès et se sont révélées saines un an plus tard. Les cartes d'anisotropie fractionnelle et de diffusivité radiale ont montré une possible myélinisation dans la région de la greffe. Les cellules souches pluripotentes induites , les cellules précurseurs d'oligodendrocytes, la correction génétique et la transplantation pour favoriser la maturation, la survie et la myélinisation des oligodendrocytes semblent être les principales voies de traitement possibles.
Pour trois types de leucodystrophies ( adrénoleucodystrophie liée à l'X (X-ALD), leucodystrophie métachromatique (MLD) et maladie de Krabbe (leucodystrophie à cellules globoïdes - GLD), la thérapie génique utilisant des cellules souches hématopoïétiques autologues pour transférer la copie saine du gène responsable de la maladie avec des vecteurs lentiviraux s'est avérée efficace et a été utilisée dans des essais cliniques pour l'X-ALD et la MLD. La progression de l'X-ALD s'est avérée perturbée par la thérapie génique par cellules souches hématopoïétiques, bien que la cause proximale de l'arrêt de la démyélinisation et la quantité de cellules souches nécessaires ne soient pas claires. Bien qu'il continue d'y avoir une accumulation d' acides gras à très longue chaîne dans le cerveau, cela ne semble pas être le facteur causal immédiat de la maladie, car la thérapie génique ne corrige pas l'accumulation.
Pour les leucodystrophies résultant d'une déficience en enzymes lysozymes, comme la maladie de Krabbe , la thérapie de remplacement enzymatique semble prometteuse. Cependant, l'administration d'enzymes s'avère difficile, car la barrière hémato-encéphalique limite considérablement ce qui peut passer dans le système nerveux central. Les recherches actuelles sur la thérapie génique pour la leucodystrophie métachromatique ont été examinées en mettant l'accent sur la transplantation ex vivo de cellules souches hématopoïétiques génétiquement modifiées.
Épidémiologie
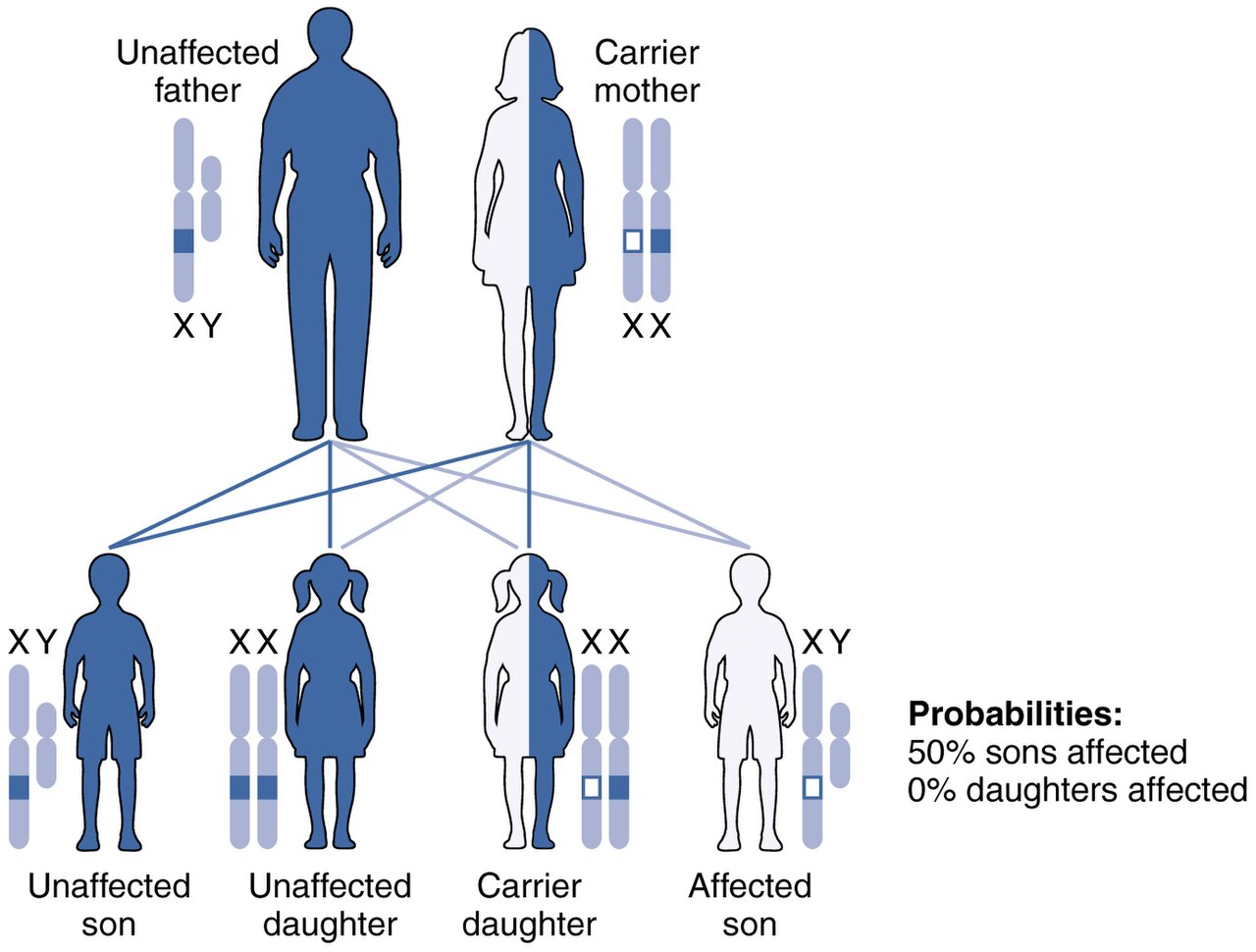
Actuellement, aucune recherche n'a montré une prévalence plus élevée de la plupart des types de leucodystrophie dans un endroit donné du monde. Il existe cependant une prévalence plus élevée de la maladie de Canavan dans la population juive. Un individu sur 40 d' origine juive ashkénaze est porteur de la maladie de Canavan. Cela représente environ 2,5 %. De plus, en raison de leurs modes de transmission autosomique récessive, il n'y a pas de différence significative entre les hommes et les femmes pour la plupart des types de leucodystrophie, y compris, mais sans s'y limiter, la leucodystrophie métachromatique, la maladie de Krabbe, la maladie de Canavan et la maladie d'Alexander. La seule exception à cette règle est tout type de leucodystrophie porté sur un chromosome sexuel , comme l'adrénoleucodystrophie liée à l'X, qui est portée sur le chromosome X. En raison du mode de transmission des maladies liées à l'X, les hommes sont plus souvent touchés par ce type de leucodystrophie, tandis que les femmes porteuses sont souvent symptomatiques, mais pas aussi gravement touchées que les hommes.
Recherche
Le National Institute of Neurological Disorders and Stroke (NINDS, sous l'égide des National Institutes of Health des États-Unis ) soutient la recherche sur les troubles génétiques, notamment les leucodystrophies. Le NINDS soutient également les chercheurs qui travaillent avec le Global Leukodystrophy Initiative Clinical Trials Network (GLIA-CTN), qui promeut les avancées dans le diagnostic et le traitement des leucodystrophies.
L'Association européenne des leucodystrophies soutient également la recherche sur les leucodystrophies. En 2020, plus de 387 projets de recherche ont été financés. Chaque année, l'ELA invite la communauté scientifique internationale à soumettre des projets de recherche dans le domaine des leucodystrophies génétiques, de la substance blanche cérébrale chez les prématurés et de la réparation de la myéline.
Société
La United Leukodystrophy Foundation (ULF), constituée en 1982, est une organisation de santé bénévole à but non lucratif qui se consacre au financement de la recherche de pointe et à la fourniture aux patients et à leurs familles d'informations sur les maladies et d'orientations médicales.
Cure MLD est un réseau mondial de défenseurs des patients et d'organismes à but non lucratif qui se consacrent à aider les familles touchées par la leucodystrophie métachromatique (MLD).
La Fondation MLD a été cofondée par Dean et Teryn Suhr en 2001 après le diagnostic de MLD en 1995 chez deux de leurs filles. La Fondation MLD sert les familles et travaille avec des chercheurs, des cliniciens, des régulateurs, des payeurs et des décideurs politiques du monde entier sur les questions liées à la MLD, à la leucodystrophie, aux maladies lysosomales et aux maladies rares.
La Leukodystrophy Alliance s'efforce de promouvoir la sensibilisation et la qualité des soins pour les personnes atteintes de leucodystrophie.
Jill Kelly et son mari, le quart-arrière de la NFL Jim Kelly , ont fondé la Hunter's Hope Foundation pour financer la recherche après que leur fils Hunter (1997-2005) ait reçu un diagnostic de leucodystrophie infantile de Krabbe.
Matthew et Michael Clark, de Hull , au Royaume-Uni, souffraient de cette maladie. Ils sont tous deux décédés, respectivement en 2013 et 2016. Leur histoire a fait l'objet du documentaire de Channel 4 The Curious Case of the Clark Brothers .
Augusto et Michaela Odone ont fondé le Myelin Project après que leur fils Lorenzo a été diagnostiqué d'adrénoleucodystrophie (ALD). Le film de 1992, Lorenzo's Oil, est une histoire vraie sur un garçon atteint d'adrénoleucodystrophie (ALD).