Dans la prédiction de la structure des protéines , les potentiels statistiques ou les potentiels basés sur les connaissances sont des fonctions de notation dérivées d'une analyse des structures protéiques connues dans la Protein Data Bank (PDB).
La méthode originale pour obtenir de tels potentiels est l' approximation quasi-chimique , due à Miyazawa et Jernigan. Elle a été suivie plus tard par le potentiel de force moyenne (PMF statistique ), développé par Sippl. Bien que les scores obtenus soient souvent considérés comme des approximations de l' énergie libre — donc appelés pseudo-énergies — cette interprétation physique est incorrecte. Néanmoins, ils sont appliqués avec succès dans de nombreux cas, car ils sont fréquemment corrélés avec les différences réelles d'énergie libre de Gibbs .
Aperçu
Les caractéristiques possibles auxquelles une pseudo-énergie peut être attribuée incluent :
- distances interatomiques ,
- angles de torsion ,
- exposition aux solvants ,
- ou géométrie de liaison hydrogène .
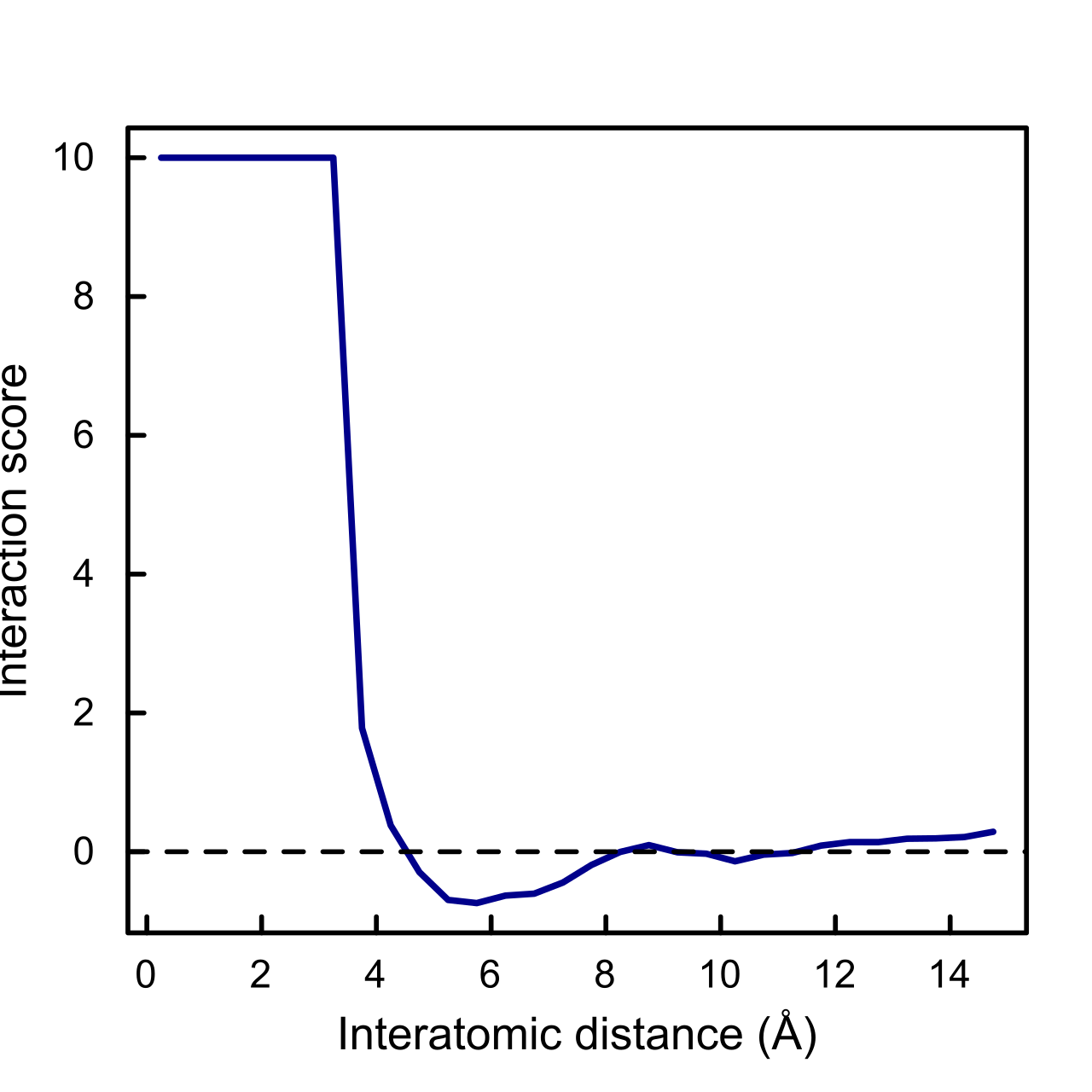
L'application classique repose cependant sur les contacts ou les distances entre acides aminés par paires , ce qui produit des potentiels interatomiques statistiques . Pour les contacts entre acides aminés par paires, un potentiel statistique est formulé sous forme de matrice d'interaction qui attribue un poids ou une valeur énergétique à chaque paire possible d' acides aminés standard . L'énergie d'un modèle structurel particulier est alors l'énergie combinée de tous les contacts par paires (définis comme deux acides aminés à une certaine distance l'un de l'autre) dans la structure. Les énergies sont déterminées à l'aide de statistiques sur les contacts entre acides aminés dans une base de données de structures protéiques connues (obtenue à partir de la PDB ).
Histoire
Développement initial
De nombreux manuels présentent les PMF statistiques proposées par Sippl comme une simple conséquence de la distribution de Boltzmann , appliquée aux distances par paires entre acides aminés. C'est incorrect, mais c'est un bon début pour introduire la construction du potentiel dans la pratique. La distribution de Boltzmann appliquée à une paire spécifique d'acides aminés est donnée par :
où est la distance, est la constante de Boltzmann , est la température et est la fonction de partition , avec
La quantité est l'énergie libre attribuée au système par paires. Un simple réarrangement conduit à la formule de Boltzmann inverse , qui exprime l'énergie libre en fonction de :
Pour construire un PMF, on introduit ensuite un état dit de référence avec une fonction de distribution et de partition correspondante , et on calcule la différence d'énergie libre suivante :
L'état de référence résulte généralement d'un système hypothétique dans lequel les interactions spécifiques entre les acides aminés sont absentes. Le second terme impliquant et peut être ignoré, car il s'agit d'une constante.
En pratique, est estimée à partir de la base de données des structures protéiques connues, tandis que résulte généralement de calculs ou de simulations. Par exemple, pourrait être la probabilité conditionnelle de trouver les atomes d'une valine et d'une sérine à une distance donnée l'un de l'autre, donnant lieu à la différence d'énergie libre . La différence totale d'énergie libre d'une protéine, , est alors considérée comme la somme de toutes les énergies libres par paires :
où la somme s'étend sur toutes les paires d'acides aminés (avec ) et est leur distance correspondante. Dans de nombreuses études, elle ne dépend pas de la séquence d'acides aminés .
Questions conceptuelles
Intuitivement, il est clair qu'une faible valeur de indique que l'ensemble des distances dans une structure est plus probable dans les protéines que dans l'état de référence. Cependant, la signification physique de ces PMF statistiques a été largement contestée depuis leur introduction. Les principaux problèmes sont les suivants :
- L'interprétation erronée de ce « potentiel » comme un véritable potentiel de force moyenne physiquement valide ;
- La nature de l’ état dit de référence et sa formulation optimale ;
- La validité des généralisations au-delà des distances par paires.
Analogie controversée
En réponse à la question de la validité physique, la première justification des PMF statistiques a été tentée par Sippl. Elle était basée sur une analogie avec la physique statistique des liquides. Pour les liquides, le potentiel de force moyenne est lié à la fonction de distribution radiale , qui est donnée par :
où et sont les probabilités respectives de trouver deux particules à distance l'une de l'autre dans le liquide et dans l'état de référence. Pour les liquides, l'état de référence est clairement défini ; il correspond au gaz idéal, constitué de particules non interactives. Le potentiel de force moyenne à deux particules est lié à par :
Selon le théorème du travail réversible, le potentiel à deux particules de force moyenne est le travail réversible nécessaire pour amener deux particules dans le liquide d'une séparation infinie à une certaine distance l'une de l'autre.
Sippl a justifié l'utilisation des PMF statistiques, quelques années après les avoir introduites pour la prédiction de la structure des protéines, en faisant appel à l'analogie avec le théorème du travail réversible pour les liquides. Pour les liquides, peut être mesuré expérimentalement en utilisant la diffusion des rayons X aux petits angles ; pour les protéines, est obtenu à partir de l'ensemble des structures protéiques connues, comme expliqué dans la section précédente. Cependant, comme l'a écrit Ben-Naim dans une publication sur le sujet :
[...] les quantités, appelées « potentiels statistiques », « potentiels basés sur la structure » ou « potentiels de paire de force moyenne », telles qu'elles sont dérivées de la banque de données sur les protéines (PDB), ne sont ni des « potentiels » ni des « potentiels de force moyenne », au sens ordinaire utilisé dans la littérature sur les liquides et les solutions.
De plus, cette analogie ne résout pas la question de savoir comment spécifier un état de référence approprié pour les protéines.
Apprentissage automatique
Au milieu des années 2000, les auteurs ont commencé à combiner plusieurs potentiels statistiques, dérivés de différentes caractéristiques structurelles, dans des scores composites . À cette fin, ils ont utilisé des techniques d'apprentissage automatique , telles que les machines à vecteurs de support (SVM). Les réseaux neuronaux probabilistes (PNN) ont également été appliqués à l'entraînement d'un potentiel statistique dépendant de la distance et de la position. En 2016, le laboratoire de recherche en intelligence artificielle DeepMind a commencé à appliquer des techniques d'apprentissage profond au développement d'un potentiel statistique dépendant de la torsion et de la distance. La méthode résultante, nommée AlphaFold , a remporté la 13e évaluation critique des techniques de prédiction de la structure des protéines (CASP) en prédisant correctement la structure la plus précise pour 25 des 43 domaines de modélisation libres .
Explication
Probabilité bayésienne
Baker et ses collègues ont justifié les PMF statistiques d'un point de vue bayésien et ont utilisé ces connaissances dans la construction de la fonction d'énergie ROSETTA à gros grains . Selon le calcul des probabilités bayésien , la probabilité conditionnelle d'une structure , étant donné la séquence d'acides aminés , peut s'écrire comme suit :
où le produit court sur toutes les paires d'acides aminés (avec ), et est la distance entre les acides aminés et . Évidemment, l'opposé du logarithme de l'expression a la même forme fonctionnelle que les PMF statistiques classiques de distance par paires, le dénominateur jouant le rôle d'état de référence. Cette explication a deux défauts : elle repose sur l'hypothèse non fondée selon laquelle la vraisemblance peut être exprimée comme un produit de probabilités par paires, et elle est purement qualitative .
Cinématique probabiliste
Hamelryck et ses collègues ont donné plus tard une explication quantitative des potentiels statistiques, selon laquelle ils se rapprochent d'une forme de raisonnement probabiliste due à Richard Jeffrey et nommée cinématique de probabilité . Cette variante de la pensée bayésienne (parfois appelée « conditionnement de Jeffrey ») permet de mettre à jour une distribution a priori en fonction de nouvelles informations sur les probabilités des éléments d'une partition sur le support de la distribution a priori. De ce point de vue, (i) il n'est pas nécessaire de supposer que la base de données des structures protéiques - utilisée pour construire les potentiels - suit une distribution de Boltzmann, (ii) les potentiels statistiques se généralisent facilement au-delà des différences par paires, et (iii) le rapport de référence est déterminé par la distribution a priori.
Ratio de référence
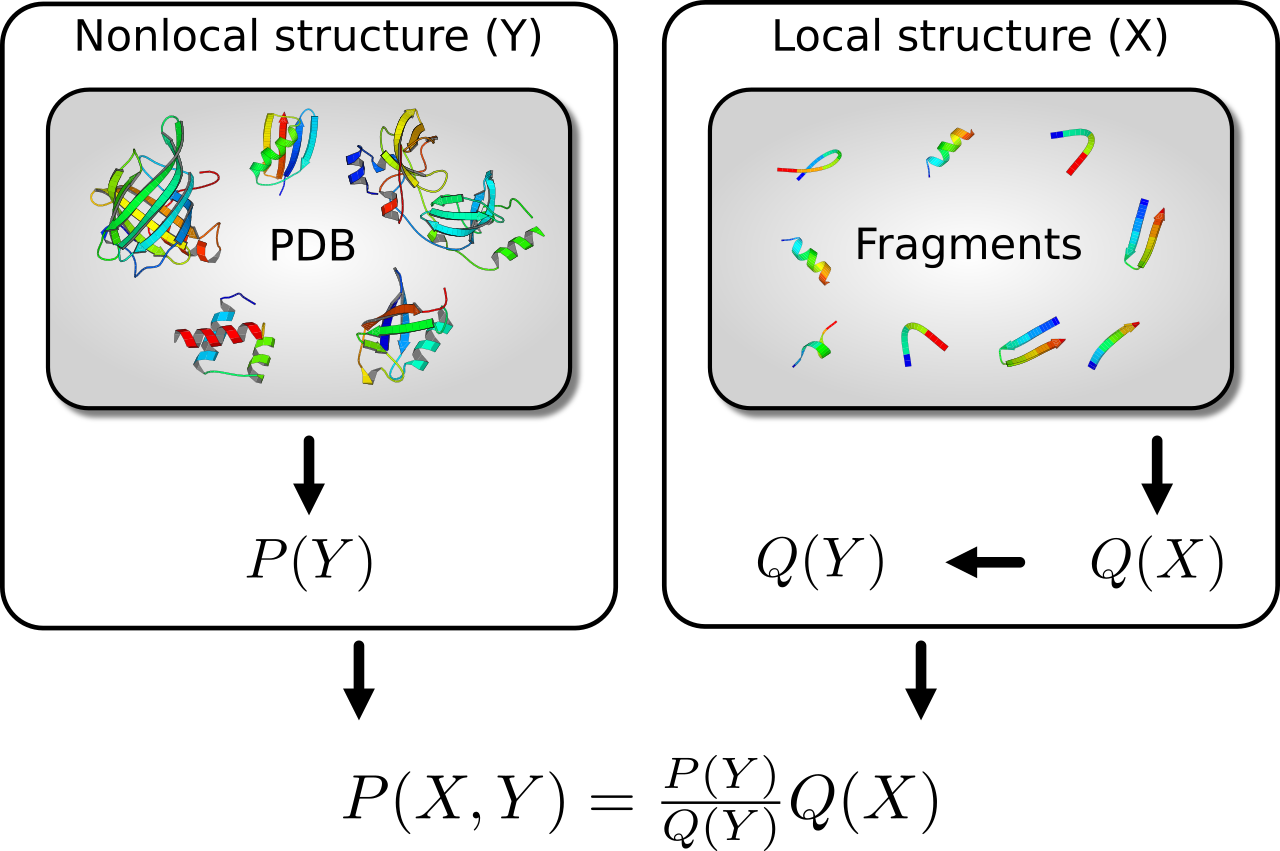
Les expressions qui ressemblent aux PMF statistiques résultent naturellement de l'application de la théorie des probabilités pour résoudre un problème fondamental qui se pose dans la prédiction de la structure des protéines : comment améliorer une distribution de probabilité imparfaite sur une première variable en utilisant une distribution de probabilité sur une seconde variable , avec . En général, et sont respectivement des variables à granularité fine et grossière. Par exemple, pourrait concerner la structure locale de la protéine, tandis que pourrait concerner les distances par paires entre les acides aminés. Dans ce cas, pourrait par exemple être un vecteur d'angles dièdres qui spécifie toutes les positions des atomes (en supposant des longueurs et des angles de liaison idéaux). Afin de combiner les deux distributions, de telle sorte que la structure locale soit distribuée selon , tandis que les distances par paires soient distribuées selon , l'expression suivante est nécessaire :
où est la distribution sur impliquée par . Le ratio dans l'expression correspond à la PMF. En général, est introduit par échantillonnage (généralement à partir d'une bibliothèque de fragments) et n'est pas explicitement évalué ; le ratio, qui au contraire est explicitement évalué, correspond à la PMF de Sippl. Cette explication est quantitative et permet la généralisation des PMF statistiques des distances par paires à des variables arbitraires à gros grains. Elle fournit également une définition rigoureuse de l'état de référence, qui est impliqué par . Les applications conventionnelles des PMF statistiques à distance par paires manquent généralement de deux caractéristiques nécessaires pour les rendre totalement rigoureuses : l'utilisation d'une distribution de probabilité appropriée sur les distances par paires dans les protéines et la reconnaissance que l'état de référence est rigoureusement défini par .
Applications
Les potentiels statistiques sont utilisés comme fonctions énergétiques dans l'évaluation d'un ensemble de modèles structurels produits par modélisation d'homologie ou par enfilage de protéines . De nombreux potentiels statistiques paramétrés différemment ont montré qu'ils pouvaient identifier avec succès la structure de l'état natif à partir d'un ensemble de structures leurres ou non natives. Les potentiels statistiques ne sont pas seulement utilisés pour la prédiction de la structure des protéines , mais également pour la modélisation de la voie de repliement des protéines .