Génétique La connaissance des séquences d'ADN est devenue indispensable à la recherche biologique fondamentale, aux projets de génographie de l'ADN et à de nombreux domaines appliqués tels que le diagnostic médical , la biotechnologie , la biologie forensique , la virologie et la systématique biologique . La comparaison des séquences d'ADN saines et mutées permet de diagnostiquer différentes maladies, notamment divers cancers , de caractériser le répertoire d'anticorps et d'orienter le traitement des patients . Un séquençage rapide de l'ADN permet une prise en charge médicale plus rapide et personnalisée, ainsi que l'identification et le catalogage d'un plus grand nombre d'organismes Les progrès rapides des technologies de séquençage de l'ADN ont joué un rôle crucial dans le séquençage complet des génomes humains, ainsi que de nombreuses espèces animales, végétales et microbiennes. Les premières séquences d'ADN ont été obtenues au début des années 1970 par des chercheurs universitaires utilisant des méthodes laborieuses basées sur la chromatographie bidimensionnelle . Suite au développement des méthodes de séquençage par fluorescence avec un séquenceur d'ADN , le séquençage de l'ADN est devenu plus facile et beaucoup plus rapide Lors de l' épidémie de grippe aviaire de 1997 , le séquençage viral a permis de déterminer que le sous-type de grippe provenait d'un réassortiment génétique entre les cailles et la volaille. Cette découverte a conduit à l'adoption d'une législation à Hong Kong interdisant la vente simultanée de cailles et de volailles vivantes sur les marchés. Le séquençage viral peut également être utilisé pour estimer la date de début d'une épidémie virale grâce à une technique d'horloge moléculaire . Les techniciens médicaux peuvent séquencer les gènes (ou, en théorie, le génome complet) des patients afin de déterminer s'il existe un risque de maladies génétiques. Il s'agit d'une forme de test génétique , bien que tous les tests génétiques n'impliquent pas le séquençage complet de l'ADN du génome. Depuis 2013, le séquençage de l'ADN est de plus en plus utilisé pour diagnostiquer et traiter les maladies rares. À mesure que de nouveaux gènes responsables de maladies génétiques rares sont identifiés, le diagnostic moléculaire devient une pratique courante. Le séquençage de l'ADN permet aux cliniciens d'identifier les maladies génétiques, d'améliorer leur prise en charge, de proposer des conseils en matière de reproduction et de mettre en œuvre des thérapies plus efficaces. Les panels de séquençage génique sont utilisés pour identifier plusieurs causes génétiques potentielles d'une maladie suspectée. De plus, le séquençage de l'ADN peut s'avérer utile pour identifier une bactérie spécifique, permettant ainsi des traitements antibiotiques plus précis et réduisant de ce fait le risque d'apparition de résistances aux antimicrobiens au sein des populations bactériennes. Chez presque tous les organismes, l'ADN est synthétisé in vivo en utilisant uniquement les 4 bases canoniques ; les modifications qui surviennent après la réplication créent d'autres bases comme la 5-méthylcytosine. Cependant, certains bactériophages peuvent incorporer directement une base non standard. Outre les modifications, l’ADN est constamment agressé par des agents environnementaux tels que les UV et les radicaux libres d’oxygène. À l’heure actuelle, la présence de telles bases endommagées n’est pas détectée par la plupart des méthodes de séquençage de l’ADN, bien que PacBio ait publié à ce sujet. L'acide désoxyribonucléique ( ADN ) a été découvert et isolé pour la première fois par Friedrich Miescher en 1869, mais il est resté peu étudié pendant des décennies car on pensait que c'étaient les protéines , et non l'ADN, qui détenaient le plan génétique de la vie. Cette situation a changé après 1944 grâce aux expériences d' Oswald Avery , Colin MacLeod et Maclyn McCarty, qui ont démontré que l'ADN purifié pouvait transformer une souche bactérienne en une autre. C'était la première fois qu'il était démontré que l'ADN était capable de modifier les propriétés des cellules.James Watson et Francis Crick ont proposé leur modèle de l'ADN en double hélice , basé sur les structures cristallisées aux rayons X étudiées par Rosalind Franklin . Selon ce modèle, l'ADN est composé de deux brins de nucléotides enroulés l'un autour de l'autre, liés par des liaisons hydrogène et orientés en sens opposé. Chaque brin est composé de quatre nucléotides complémentaires – adénine (A), cytosine (C), guanine (G) et thymine (T) – l'adénine (A) d'un brin étant toujours appariée à la thymine (T) de l'autre, et la cytosine (C) toujours appariée à la guanine (G). Ils ont suggéré qu'une telle structure permettait à chaque brin de reconstruire l'autre, une idée fondamentale pour la transmission de l'information héréditaire d'une génération à l'autre. Les travaux de Frederick Sanger ont posé les bases du séquençage des protéines . En 1955, il avait achevé le séquençage de tous les acides aminés de l'insuline , une petite protéine sécrétée par le pancréas. Cette découverte a apporté la première preuve concluante que les protéines étaient des entités chimiques dotées d'une structure moléculaire spécifique, et non un mélange aléatoire de substances en suspension dans un fluide. Le succès de Sanger dans le séquençage de l'insuline a stimulé les cristallographes aux rayons X, notamment Watson et Crick, qui cherchaient alors à comprendre comment l'ADN dirigeait la formation des protéines au sein de la cellule. Peu après avoir assisté à une série de conférences données par Frederick Sanger en octobre 1954, Crick a commencé à élaborer une théorie selon laquelle l'agencement des nucléotides dans l'ADN déterminait la séquence des acides aminés dans les protéines, ce qui, à son tour, contribuait à déterminer la fonction d'une protéine. Il a publié cette théorie en 1958. Le séquençage de l'ARN fut l'une des premières formes de séquençage de nucléotides. Son avancée majeure réside dans le séquençage du premier gène complet et du génome complet du bactériophage MS2 , identifiés et publiés par Walter Fiers et ses collaborateurs de l' Université de Gand ( Gand , Belgique ), en 1972 et 1976 Les méthodes traditionnelles de séquençage de l'ARN nécessitent la synthèse d'une molécule d'ADNc qui doit ensuite être séquencée La première méthode de détermination des séquences d'ADN reposait sur une stratégie d'extension d'amorces spécifiques de localisation, mise au point par le généticien Ray Wu à l'Université Cornell en 1970. La catalyse par l'ADN polymérase et le marquage spécifique des nucléotides, deux techniques essentielles des méthodes de séquençage actuelles, ont été utilisés pour séquencer les extrémités cohésives de l'ADN du phage lambda . Entre 1970 et 1973, Wu, la scientifique Radha Padmanabhan et leurs collègues ont démontré que cette méthode pouvait être employée pour déterminer n'importe quelle séquence d'ADN à l'aide d'amorces synthétiques spécifiques de localisation. Walter Gilbert , biochimiste, et Allan Maxam , généticien moléculaire, ont également mis au point à Harvard des méthodes de séquençage, dont une pour le « séquençage de l’ADN par dégradation chimique » . En 1973, Gilbert et Maxam ont publié la séquence de 24 paires de bases grâce à une méthode appelée analyse des spots errants . Les progrès en matière de séquençage ont été favorisés par le développement concomitant de la technologie de l’ADN recombinant , permettant d’isoler des échantillons d’ADN à partir de sources autres que les virus Deux ans plus tard, en 1975, Frederick Sanger , biochimiste, et Alan Coulson , généticien, ont mis au point une méthode de séquençage de l'ADN. Cette technique, dite « méthode Plus et Moins », consistait à fournir tous les composants de l'ADN, mais à exclure la réaction de l'une des quatre bases nécessaires à la synthèse complète de l'ADN. En 1976, Gilbert et Maxam ont inventé à Harvard une méthode de séquençage rapide de l'ADN, connue sous le nom de séquençage Maxam-Gilbert. Cette technique consiste à traiter l'ADN radiomarqué avec un produit chimique et à utiliser un gel de polyacrylamide pour déterminer la séquence. En 1977, Sanger adopta une stratégie d'extension d'amorces pour développer des méthodes de séquençage d'ADN plus rapides au Centre MRC de Cambridge , au Royaume-Uni. Cette technique était similaire à sa stratégie « Plus et Moins », mais elle reposait sur l'incorporation sélective de didésoxynucléotides (ddNTP) terminaux par l'ADN polymérase lors de la réplication d'ADN in vitro . Sanger publia cette méthode la même année. Une méthode non radioactive de transfert des molécules d'ADN des mélanges de réaction de séquençage sur une matrice d'immobilisation pendant l'électrophorèse a été développée par Herbert Pohl et ses collaborateurs au début des années 1980. Suite à la commercialisation du séquenceur d'ADN « Direct-Blotting-Electrophoresis-System GATC 1500 » par GATC Biotech , qui a été intensivement utilisé dans le cadre du programme de séquençage du génome de l'UE, la séquence complète d'ADN du chromosome II de la levure Saccharomyces cerevisiae . En 1986, le laboratoire de Leroy E. Hood au California Institute of Technology annonça la mise au point du premier séquenceur d'ADN semi-automatisé. Applied Biosystems commercialisa ensuite, en 1987, le premier séquenceur entièrement automatisé, l'ABI 370, puis Dupont lança le Genesis 2000 , qui utilisait une nouvelle technique de marquage fluorescent permettant l'identification des quatre didésoxynucléotides sur une seule voie. Dès 1990, les National Institutes of Health (NIH) américains entreprirent des essais de séquençage à grande échelle sur Mycoplasma capricolum , Escherichia coli , Caenorhabditis elegans et Saccharomyces cerevisiae , pour un coût de 0,75 $US par base. Parallèlement, le séquençage des séquences d'ADNc humaines , appelées étiquettes de séquences exprimées (EST), a débuté au laboratoire de Craig Venter , dans le but de capturer la fraction codante du génome humain . En 1995, Venter, Hamilton Smith et leurs collègues de l'Institut de recherche génomique (TIGR) ont publié le premier génome complet d'un organisme libre, la bactérie Haemophilus influenzae . Le chromosome circulaire contient 1 830 137 paires de bases et sa publication dans la revue Science a marqué la première utilisation publiée du séquençage shotgun du génome entier, éliminant ainsi la nécessité d'efforts de cartographie initiaux. En 2003, les méthodes de séquençage aléatoire du Projet Génome Humain ont permis d'obtenir une première ébauche de la séquence du génome humain, avec une précision de 92 %. En 2022, les scientifiques ont séquencé avec succès les 8 % restants du génome humain. Le gène de référence standard entièrement séquencé est appelé GRCh38.p14 et contient 3,1 milliards de paires de bases. Le 26 octobre 1990, Roger Tsien , Pepi Ross, Margaret Fahnestock et Allan J. Johnston ont déposé un brevet décrivant un séquençage par étapes (« base par base ») avec des bloqueurs 3' amovibles sur des puces à ADN (transferts et molécules d'ADN uniques). En 1996, Pål Nyrén et son étudiant Mostafa Ronaghi, de l'Institut royal de technologie de Stockholm, ont publié leur méthode de pyroséquençage . Le 1er avril 1997, Pascal Mayer et Laurent Farinelli ont déposé des brevets décrivant le séquençage de colonies d'ADN. Les méthodes de préparation d'échantillons d'ADN et de réaction en chaîne par polymérase (PCR) aléatoires sur surface décrites dans ce brevet, couplées à la méthode de séquençage « base par base » de Roger Tsien et al., sont maintenant mises en œuvre dans les séquenceurs de génomes Hi-Seq d' Illumina .score de qualité Phred pour l'analyse des données de séquenceurs, une technique d'analyse marquante qui a été largement adoptée et qui reste la mesure la plus courante pour évaluer la précision d'une plateforme de séquençage. Lynx Therapeutics a publié et commercialisé le séquençage de signature massivement parallèle (MPSS) en 2000. Cette méthode intégrait une technologie de séquençage parallélisée, médiée par adaptateur/ligation et basée sur des billes et a servi de première méthode de séquençage « nouvelle génération » disponible commercialement, bien qu'aucun séquenceur d'ADN n'ait été vendu à des laboratoires indépendants. Cette méthode est largement obsolète depuis 2023. L'objectif du séquençage séquentiel par synthèse (SBS) est de déterminer la séquence d'un échantillon d'ADN en détectant l'incorporation d'un nucléotide par une ADN polymérase . Une polymérase modifiée est utilisée pour synthétiser une copie d'un brin d'ADN simple brin, et l'incorporation de chaque nucléotide est suivie. Le principe du séquençage en temps réel par synthèse a été décrit pour la première fois en 1993 et des améliorations ont été publiées quelques années plus tard. Les étapes clés sont très similaires pour tous les modes de réalisation du SBS et comprennent : (1) l’amplification de l’ADN (pour renforcer le signal ultérieur) et la fixation de l’ADN à séquencer sur un support solide (à l’exception du PacBio SMRT ) ; (2) la génération d’ADN simple brin sur le support solide ; (3) l’incorporation de nucléotides à l’aide d’une polymérase modifiée ; et (4) la détection en temps réel de l’incorporation des nucléotides. Les étapes 3 et 4 sont répétées et la séquence est assemblée à partir des signaux obtenus à l’étape 4. Ce principe de séquençage par synthèse en temps réel a été utilisé pour la quasi-totalité des séquenceurs à haut débit , notamment 454 , PacBio , Ion Torrent , Illumina et MGI . En février 2025, Roche a annoncé la mise au point d'une nouvelle méthode de séquençage exclusive basée sur de petites molécules. Cette méthode « encode la séquence d'une molécule d'acide nucléique cible (ADN ou ARN) dans un polymère de substitution mesurable appelé Xpandomer » et combine l'amplification avec une détection ultérieure par une plateforme nanopore équipée d'une caméra CMOS . Le séquençage à grande échelle vise souvent à séquencer de très longs fragments d'ADN, tels que des chromosomes entiers . Il peut également servir à générer un très grand nombre de courtes séquences, comme celles obtenues par la technique du phage display . Pour les cibles plus longues, telles que les chromosomes, les méthodes courantes consistent à couper (à l'aide d'enzymes de restriction ) ou à fragmenter (par des forces mécaniques) de grands fragments d'ADN en fragments plus courts. L'ADN fragmenté peut ensuite être cloné dans un vecteur d'ADN et amplifié dans une bactérie hôte comme Escherichia coli . Les courts fragments d'ADN purifiés à partir de colonies bactériennes individuelles sont séquencés individuellement et assemblés électroniquement en une longue séquence contiguë. Des études ont montré que l'ajout d'une étape de sélection de taille, afin de collecter des fragments d'ADN de taille uniforme, peut améliorer l'efficacité du séquençage et la précision de l'assemblage du génome. Dans ces études, le dimensionnement automatisé s'est avéré plus reproductible et précis que le dimensionnement manuel sur gel. Le terme « séquençage de novo » désigne spécifiquement les méthodes utilisées pour déterminer la séquence d'ADN à partir de séquences non connues. « De novo » signifie « depuis le début » en latin. Les lacunes dans la séquence assemblée peuvent être comblées par extension d'amorces . Les différentes stratégies présentent des compromis différents en termes de vitesse et de précision ; les méthodes de séquençage aléatoire sont souvent utilisées pour le séquençage de grands génomes, mais leur assemblage est complexe et difficile, notamment en raison des séquences répétées qui entraînent fréquemment des lacunes dans l'assemblage du génome.réaction en chaîne par polymérase (PCR) recouvre ensuite chaque bille de copies clonales de la molécule d'ADN, suivie d'une immobilisation pour un séquençage ultérieur. La PCR en émulsion est utilisée dans les méthodes développées par Marguilis et al. (commercialisées par 454 Life Sciences ), Shendure et Porreca et al. (également connue sous le nom de « séquençage polony ») et le séquençage SOLiD (développé par Agencourt , puis Applied Biosystems , et maintenant Life Technologies ) . La PCR en émulsion est également utilisée dans les plateformes GemCode et Chromium développées par 10x Genomics . Le séquençage à haut débit, qui comprend les méthodes de séquençage de nouvelle génération « à lecture courte » et de troisième génération « à lecture longue », s'applique au séquençage de l'exome , au séquençage du génome, au reséquençage du génome, au profilage du transcriptome ( RNA-Seq ), aux interactions ADN-protéines ( ChIP-sequencing ) et à la caractérisation de l'épigénome . La forte demande de séquençage à faible coût a stimulé le développement de technologies de séquençage à haut débit qui parallélisent le processus de séquençage, produisant simultanément des milliers, voire des millions de séquences. Ces technologies visent à réduire le coût du séquençage de l'ADN au-delà des limites des méthodes classiques de marquage par colorant. Le séquençage à ultra-haut débit permet d'exécuter jusqu'à 500 000 opérations de séquençage par synthèse en parallèle. Ces technologies ont permis de séquencer l'intégralité du génome humain en une seule journée. Illumina , Qiagen et ThermoFisher Scientific . longueur de lecture maximale >100 000 bases MiSeq : 50–600 pb ; HiSeq 2500 : 50–500 pb ; HiSeq 3/4000 : 50–300 pb ; HiSeq X : 300 pb NextSeq : 130-00 millions ; HiSeq 2500 : 300 millions – 2 milliards ; HiSeq 3/4000 2,5 milliards ; HiSeq X : 3 milliards MGISEQ 200 : 50-200 pb ; BGISEQ-500, MGISEQ-2000 : 50-300 pb MGISEQ 200 : 300 M ; BGISEQ-500 : 1300 M par cellule d’écoulement ; MGISEQ-2000 : cellule d’écoulement FCS de 375 M, cellule d’écoulement FCL de 1 500 M par cellule d’écoulement. Le concept repose sur l'idée que des molécules d'ADN ou d'ARN simple brin peuvent être entraînées par électrophorèse selon une séquence linéaire stricte à travers un pore biologique de moins de huit nanomètres, et détectées grâce au courant ionique qu'elles génèrent lors de leur passage. Le pore contient une région de détection capable de reconnaître différentes bases, chaque base générant des signaux temporels spécifiques correspondant à la séquence de bases traversant le pore, lesquels sont ensuite analysés. Un contrôle précis du transport de l'ADN à travers le pore est essentiel au succès de l'expérience. Différentes enzymes, telles que les exonucléases et les polymérases, ont été utilisées pour moduler ce processus en étant positionnées à proximité de l'entrée du pore. Dans cette méthode, les molécules d'ADN et les amorces sont d'abord fixées sur une lame ou dans une cellule de flux, puis amplifiées par polymérase afin de former des colonies d'ADN clonales locales, appelées ultérieurement « clusters d'ADN ». Pour déterminer la séquence, quatre types de bases de terminaison réversibles (bases RT) sont ajoutés et les nucléotides non incorporés sont éliminés par lavage. Une caméra capture des images des nucléotides marqués par fluorescence . Le colorant, ainsi que le bloqueur 3' terminal, sont ensuite éliminés chimiquement de l'ADN, permettant ainsi le démarrage du cycle suivant. Contrairement au pyroséquençage, les chaînes d'ADN sont allongées nucléotide par nucléotide et l'acquisition d'images peut être différée, ce qui permet de capturer de très grands réseaux de colonies d'ADN par imagerie séquentielle à l'aide d'une seule caméra. Le découplage de la réaction enzymatique et de l'acquisition d'images permet un débit optimal et une capacité de séquençage théoriquement illimitée. Avec une configuration optimale, le débit maximal de l'instrument est ainsi déterminé uniquement par la fréquence de conversion analogique-numérique de la caméra, multipliée par le nombre de caméras et divisée par le nombre de pixels par colonie d'ADN requis pour une visualisation optimale (environ 10 pixels/colonie). En 2012, grâce à des caméras fonctionnant à des fréquences de conversion A/D supérieures à 10 MHz et aux technologies optiques, fluidiques et enzymatiques disponibles, le débit pouvait atteindre plusieurs millions de nucléotides par seconde, correspondant approximativement à un génome humain par heure et par instrument avec une couverture de 1x , et à un génome humain reséquencé (à environ 30x) par jour et par instrument (équipé d'une seule caméra). Cette méthode est une version améliorée de la technologie de ligation d'ancres de sondes combinatoires (cPAL) décrite par Complete Genomics qui a depuis été intégrée à la société chinoise de génomique BGI en 2013 Les deux entreprises ont perfectionné cette technologie afin d'obtenir des séquences plus longues, de réduire le temps de réaction et d'accélérer l'obtention des résultats. De plus, les données sont désormais générées sous forme de séquences complètes contiguës au format FASTQ standard et peuvent être utilisées telles quelles dans la plupart des chaînes de traitement bioinformatiques basées sur le séquençage à courtes lectures Les deux technologies à la base de cette technologie de séquençage à haut débit sont les nanobilles d'ADN (DNB) et les matrices structurées permettant la fixation des nanobilles sur une surface solide. Les nanobilles d'ADN sont formées par dénaturation de banques d'ADN double brin auxquelles des adaptateurs ont été ligaturés, puis par ligation du brin sens uniquement à un oligonucléotide d'amorçage pour former un cercle d'ADN simple brin (ssADN). Des copies fidèles de ces cercles contenant l'insert d'ADN sont produites par amplification en cercle roulant (RCA), générant environ 300 à 500 copies. Le long brin d'ADN simple brin se replie sur lui-même pour former une structure tridimensionnelle de nanobille d'environ 220 nm de diamètre. La fabrication des DNB remplace la nécessité de générer des copies PCR de la banque sur la cellule de flux et permet ainsi d'éliminer une grande partie des lectures dupliquées, des ligations adaptateur-adaptateur et des erreurs induites par la PCR. Un réseau structuré de points chargés positivement est fabriqué par photolithographie et gravure, suivies d'une modification chimique, afin de générer une cellule de séquençage. Chaque point de la cellule mesure environ 250 nm de diamètre et est séparé par 700 nm (d'axe en axe), ce qui facilite la fixation d'une molécule de DNB chargée négativement et réduit ainsi les risques de sous-agrégation ou de sur-agrégation. Le séquençage est ensuite réalisé par l'ajout d'une sonde oligonucléotidique qui se fixe à des sites spécifiques au sein du DNB. Cette sonde sert d'ancre, permettant la liaison de l'un des quatre nucléotides marqués, réversibles et inactivés, après son passage dans la cellule de flux. Les nucléotides non liés sont éliminés par lavage avant l'excitation laser des marqueurs fixés, qui émettent alors une fluorescence. Le signal est capturé par des caméras et converti en un signal numérique pour l'identification des bases. À la fin du cycle, le terminateur et le marqueur de la base fixée sont clivés chimiquement. Le cycle est répété avec un nouveau flux de nucléotides marqués libres traversant la cellule de flux, permettant ainsi la liaison du nucléotide suivant et la capture de son signal. Ce processus est répété plusieurs fois (généralement de 50 à 300 fois) afin de déterminer la séquence du fragment d'ADN inséré, à un débit d'environ 40 millions de nucléotides par seconde (données de 2018). La technologie SOLiD d' Applied Biosystems (désormais une marque de Life Technologies ) utilise le séquençage par ligation . Un ensemble de tous les oligonucléotides possibles de longueur fixe est marqué en fonction de la position à séquencer. Les oligonucléotides sont hybridés puis ligaturés ; la ligation préférentielle par l'ADN ligase pour les séquences complémentaires génère un signal informatif sur le nucléotide à cette position. Chaque base de la matrice est séquencée deux fois, et les données résultantes sont décodées selon le schéma de codage à 2 bases utilisé dans cette méthode. Avant le séquençage, l'ADN est amplifié par PCR en émulsion. Les billes résultantes, contenant chacune une seule copie de la même molécule d'ADN, sont déposées sur une lame de verre. On obtient ainsi des séquences en quantités et longueurs comparables à celles obtenues par séquençage Illumina. Cette méthode de séquençage par ligation présente des difficultés pour le séquençage des séquences palindromiques. Le séquençage Heliscope est une méthode de séquençage de molécules uniques développée par Helicos Biosciences . Elle utilise des fragments d'ADN auxquels ont été ajoutés des adaptateurs de queue poly-A, fixés à la surface de la cellule de flux. Les étapes suivantes consistent en un séquençage par extension, avec des lavages cycliques de la cellule de flux à l'aide de nucléotides marqués par fluorescence (un type de nucléotide à la fois, comme dans la méthode Sanger). Les lectures sont effectuées par le séquenceur Heliscope. Les séquences sont courtes, d'une longueur moyenne de 35 pb. La principale innovation de cette technologie réside dans le fait qu'elle était la première de sa catégorie à séquencer de l'ADN non amplifié, évitant ainsi les erreurs de lecture liées aux étapes d'amplification. En 2009, le génome humain a été séquencé à l'aide de l'Heliscope, mais la société a fait faillite en 2012. Deux principaux systèmes microfluidiques sont utilisés pour le séquençage de l'ADN : la microfluidique à gouttelettes et la microfluidique numérique . Les dispositifs microfluidiques permettent de surmonter de nombreuses limitations des puces de séquençage actuelles.polydiméthylsiloxane (PDMS) , ils utilisent la technique de transfert d'énergie par résonance de Förster (FRET) pour lire les séquences d'ADN contenues dans les gouttelettes. Chaque position sur la matrice est testée pour une séquence spécifique de 15 bases. Fair et al. ont utilisé des dispositifs microfluidiques numériques pour étudier le pyroséquençage de l'ADN . Parmi les avantages significatifs, citons la portabilité du dispositif, le volume de réactifs, la rapidité d'analyse, les possibilités de production en série et le haut débit. Cette étude a fourni une preuve de concept démontrant que les dispositifs numériques peuvent être utilisés pour le pyroséquençage ; elle incluait la synthèse, qui consiste en l'extension des enzymes et l'ajout de nucléotides marqués. Boles et al. ont également étudié le pyroséquençage sur des dispositifs microfluidiques numériques. Ils ont utilisé un dispositif d'électromouillage pour créer, mélanger et séparer des gouttelettes. Le séquençage repose sur un protocole à trois enzymes et des matrices d'ADN ancrées par des billes magnétiques. Le dispositif a été testé avec deux protocoles et a démontré une précision de 100 % d'après les niveaux bruts des pyrogrammes. Les avantages de ces dispositifs microfluidiques numériques incluent leur taille réduite, leur coût et les niveaux d'intégration fonctionnelle qu'ils permettent d'atteindre. La recherche sur le séquençage de l'ADN, utilisant la microfluidique, peut également être appliquée au séquençage de l'ARN , en utilisant des techniques microfluidiques à gouttelettes similaires, telles que la méthode inDrops. Cela montre que bon nombre de ces techniques de séquençage de l'ADN pourront être appliquées plus loin et être utilisées pour mieux comprendre les génomes et les transcriptomes.des nanopores (une méthode désormais commercialisée, mais les générations suivantes, telles que les nanopores à l'état solide, sont encore en développement) , et des techniques de microscopie, comme la microscopie à force atomique ou la microscopie électronique à transmission , utilisées pour identifier la position des nucléotides individuels au sein de longs fragments d'ADN ( > 5 000 pb) par marquage des nucléotides avec des éléments plus lourds (par exemple, des halogènes) pour une détection et un enregistrement visuels. Les technologies de troisième génération visent à augmenter le débit et à réduire le délai d'obtention des résultats et le coût en éliminant le besoin de réactifs en excès et en exploitant la processivité de l'ADN polymérase. Une autre approche utilise la mesure des courants de tunnel électrique à travers un brin d'ADN simple brin lorsqu'il se déplace dans un canal. Selon sa structure électronique, chaque base affecte différemment le courant de tunnel, permettant ainsi de différencier les bases. L'utilisation des courants tunnel a le potentiel de séquencer des ordres de grandeur plus rapidement que les méthodes de courant ionique et le séquençage de plusieurs oligomères d'ADN et micro-ARN a déjà été réalisé. Le séquençage par hybridation est une méthode non enzymatique qui utilise une puce à ADN . Un échantillon d'ADN dont la séquence doit être déterminée est marqué par fluorescence et hybridé à une puce contenant des séquences connues. Un signal d'hybridation intense provenant d'un point donné de la puce permet d'identifier sa séquence dans l'ADN à séquencer. Cette méthode de séquençage exploite les propriétés de liaison d'une banque de courtes molécules d'ADN simple brin (oligonucléotides), également appelées sondes d'ADN, pour reconstruire une séquence d'ADN cible. Les hybrides non spécifiques sont éliminés par lavage et l'ADN cible est élué. Les hybrides sont réarrangés afin de permettre la reconstruction de la séquence d'ADN. L'avantage de ce type de séquençage réside dans sa capacité à capturer un grand nombre de cibles avec une couverture homogène. Une grande quantité de réactifs et d'ADN de départ est généralement nécessaire. Cependant, l'avènement de l'hybridation en solution permet de réduire considérablement la quantité d'équipements et de réactifs requis. La spectrométrie de masse peut être utilisée pour déterminer les séquences d'ADN. La spectrométrie de masse MALDI-TOF (désorption/ionisation laser assistée par matrice – temps de vol ) a été étudiée comme alternative à l'électrophorèse sur gel pour la visualisation des fragments d'ADN. Cette méthode compare les fragments d'ADN obtenus par séquençage par terminaison de chaîne en fonction de leur masse plutôt que de leur taille. La masse de chaque nucléotide étant différente, cette différence est détectable par spectrométrie de masse. Les mutations d'un seul nucléotide dans un fragment sont plus facilement détectables par spectrométrie de masse que par électrophorèse sur gel seule. La spectrométrie de masse MALDI-TOF permet également de détecter plus facilement les différences entre les fragments d'ARN ; les chercheurs peuvent ainsi séquencer indirectement l'ADN par des méthodes basées sur la spectrométrie de masse en le convertissant préalablement en ARN. La résolution plus élevée des fragments d'ADN permise par les méthodes de spectrométrie de masse présente un intérêt particulier pour les chercheurs en sciences forensiques, car ils peuvent être amenés à identifier des individus en détectant des polymorphismes nucléotidiques simples (SNP) dans des échantillons d'ADN humain. Ces échantillons étant souvent fortement dégradés, les chercheurs en sciences forensiques privilégient généralement l'ADN mitochondrial pour sa plus grande stabilité et ses applications dans les études de lignée. Les méthodes de séquençage par spectrométrie de masse ont été utilisées pour comparer les séquences d'ADN mitochondrial humain provenant d'échantillons de la base de données du FBI et d'ossements trouvés dans des fosses communes de soldats de la Première Guerre mondiale Les premières méthodes de séquençage par terminaison de chaîne et de spectrométrie de masse à temps de vol (TOF MS) ont permis d'obtenir des séquences d'une longueur allant jusqu'à 100 paires de bases . Les chercheurs n'ont pas réussi à dépasser cette taille moyenne ; à l'instar du séquençage par terminaison de chaîne seul, le séquençage d'ADN par spectrométrie de masse pourrait ne pas convenir aux projets de séquençage de novo à grande échelle. Néanmoins, une étude de 2010 a utilisé ces courtes séquences et la spectrométrie de masse pour comparer les polymorphismes nucléotidiques simples (SNP) chez des souches de Streptococcus pathogènes . Une méthode a été mise au point pour analyser des ensembles complets d' interactions protéiques grâce à une combinaison de pyroséquençage 454 et d'une méthode d'affichage d'ARNm viral in vitro . Concrètement, cette méthode lie de manière covalente les protéines d'intérêt aux ARNm qui les codent, puis détecte les fragments d'ARNm par RT- PCR . L'ARNm peut ensuite être amplifié et séquencé. Cette méthode combinée, appelée IVV-HiTSeq, peut être mise en œuvre en milieu acellulaire, bien que ses résultats puissent ne pas être représentatifs des conditions in vivo . Bien qu’il existe de nombreuses méthodes de séquençage de l’ADN, seules quelques-unes dominent le marché. En 2022, Illumina détenait environ 80 % du marché ; le reste étant partagé par quelques acteurs seulement (PacBio, Oxford, 454, MGI) Le succès de tout protocole de séquençage d'ADN repose sur l'extraction et la préparation de l'échantillon d'ADN ou d'ARN à partir du matériel biologique d'intérêt.enzyme synthétise un ADNc double brin à partir des brins d'ARN existants, et l'ADNc ainsi obtenu peut être séquencé. Cette réaction est généralement amorcée par des hexamères aléatoires ou des amorces ciblant la queue poly(A) de l'ARNm . L'ADNc peut ensuite être traité de la même manière que l'ADN génomique. Après extraction d'ADN ou d'ARN, les échantillons peuvent nécessiter une préparation supplémentaire selon la méthode de séquençage. Pour le séquençage Sanger, un clonage ou une PCR est requis avant le séquençage. Dans le cas des méthodes de séquençage de nouvelle génération, la préparation de la librairie est nécessaire avant le traitement. L'évaluation de la qualité et de la quantité des acides nucléiques, tant après extraction qu'après préparation de la librairie, permet d'identifier les échantillons dégradés, fragmentés ou de faible pureté et d'obtenir des données de séquençage de haute qualité. En octobre 2006, la Fondation X Prize a lancé une initiative visant à promouvoir le développement des technologies de séquençage du génome complet , appelée Archon X Prize , avec l'intention d'attribuer 10 millions de dollars à « la première équipe capable de construire un appareil et de l'utiliser pour séquencer 100 génomes humains en 10 jours ou moins, avec une précision d'au plus une erreur pour 100 000 bases séquencées, les séquences couvrant avec précision au moins 98 % du génome, et à un coût récurrent n'excédant pas 10 000 dollars américains par génome ». Chaque année, le National Human Genome Research Institute (NHGRI) octroie des subventions pour de nouvelles recherches et de nouveaux développements en génomique . Les subventions de 2010 et les candidats de 2011 concernent notamment la poursuite des travaux sur les méthodologies de séquençage microfluidique, polony et à forte densité de bases. Les technologies de séquençage décrites ici produisent des données brutes qui doivent être assemblées en séquences plus longues, telles que des génomes complets ( assemblage de séquences ). De nombreux défis informatiques se posent, notamment l'évaluation des données de séquençage brutes, réalisée par des programmes et des algorithmes comme Phred et Phrap . D'autres défis consistent à gérer les séquences répétitives qui, souvent présentes à différents endroits du génome, empêchent l'assemblage complet des génomes. Par conséquent, de nombreuses séquences peuvent ne pas être assignées à des chromosomes spécifiques . La production des données de séquençage brutes ne constitue que le point de départ de leur analyse bioinformatique détaillée . De nouvelles méthodes de séquençage et de correction des erreurs de séquençage ont néanmoins été développées. Parfois, les séquences brutes produites par le séquenceur ne sont correctes et précises que sur une fraction de leur longueur. L'utilisation de la séquence complète peut introduire des artefacts dans les analyses ultérieures, telles que l'assemblage du génome, l'identification des SNP ou l'estimation de l'expression génique. Deux classes de programmes d'élagage ont été introduites : les algorithmes à fenêtre et les algorithmes de somme cumulée. Voici une liste partielle des algorithmes d'élagage actuellement disponibles, avec indication de leur classe : Avec la généralisation du séquençage de l'ADN, le stockage, la sécurité et le partage des données génomiques revêtent une importance croissante. Par exemple, on craint que les assureurs n'utilisent les données génomiques d'un individu pour modifier ses tarifs, en fonction d'une estimation de son état de santé futur basée sur son ADN. En mai 2008, la loi américaine sur la non-discrimination génétique (GINA) a été promulguée, interdisant toute discrimination fondée sur l'information génétique en matière d'assurance maladie et d'emploi. En 2012, la Commission présidentielle américaine pour l'étude des questions de bioéthique a conclu que la législation existante en matière de protection de la vie privée pour les données de séquençage de l'ADN, telle que la GINA et la loi HIPAA (Health Insurance Portability and Accountability Act) , était insuffisante. Elle a notamment souligné la sensibilité particulière des données de séquençage du génome entier, qui pourraient permettre d'identifier non seulement l'individu dont elles proviennent, mais aussi ses proches. Dans la plupart des États américains, l’ADN « abandonné », par exemple celui trouvé sur un timbre ou une enveloppe léchés, une tasse de café, une cigarette, un chewing-gum, des ordures ménagères ou des cheveux tombés sur un trottoir, peut être légalement prélevé et séquencé par quiconque, y compris la police, les enquêteurs privés, les opposants politiques ou les personnes impliquées dans des litiges de paternité. En 2013, onze États disposaient de lois pouvant être interprétées comme interdisant le « vol d’ADN ». L'utilisation croissante du dépistage des variations génétiques, tant chez les nouveau-nés que chez les adultes par des entreprises comme 23andMe , soulève également des questions éthiques . Il a été avancé que ce dépistage pouvait être néfaste, en augmentant l'anxiété chez les personnes présentant un risque accru de maladie. Par exemple, dans un cas rapporté par Time , des médecins dépistant des variants génétiques chez un nourrisson malade ont choisi de ne pas informer les parents de l'existence d'un variant sans lien avec la démence , par crainte du préjudice que cela leur causerait. Cependant, une étude de 2011 publiée dans le New England Journal of Medicine a montré que les personnes ayant subi un profilage des risques de maladie ne présentaient pas d'augmentation de leur niveau d'anxiété. Par ailleurs, le développement des technologies de séquençage de nouvelle génération, telles que le séquençage nanopore, a également soulevé de nouvelles préoccupations éthiques. 
séquence d'acide nucléique , c'est-à-dire l'ordre des nucléotides dans l'ADN . Il englobe toute méthode ou technologie utilisée pour déterminer l'ordre des quatre bases : adénine , thymine , cytosine et guanine . L'avènement des méthodes de séquençage rapide de l'ADN a considérablement accéléré la recherche et les découvertes en biologie et en médecine. 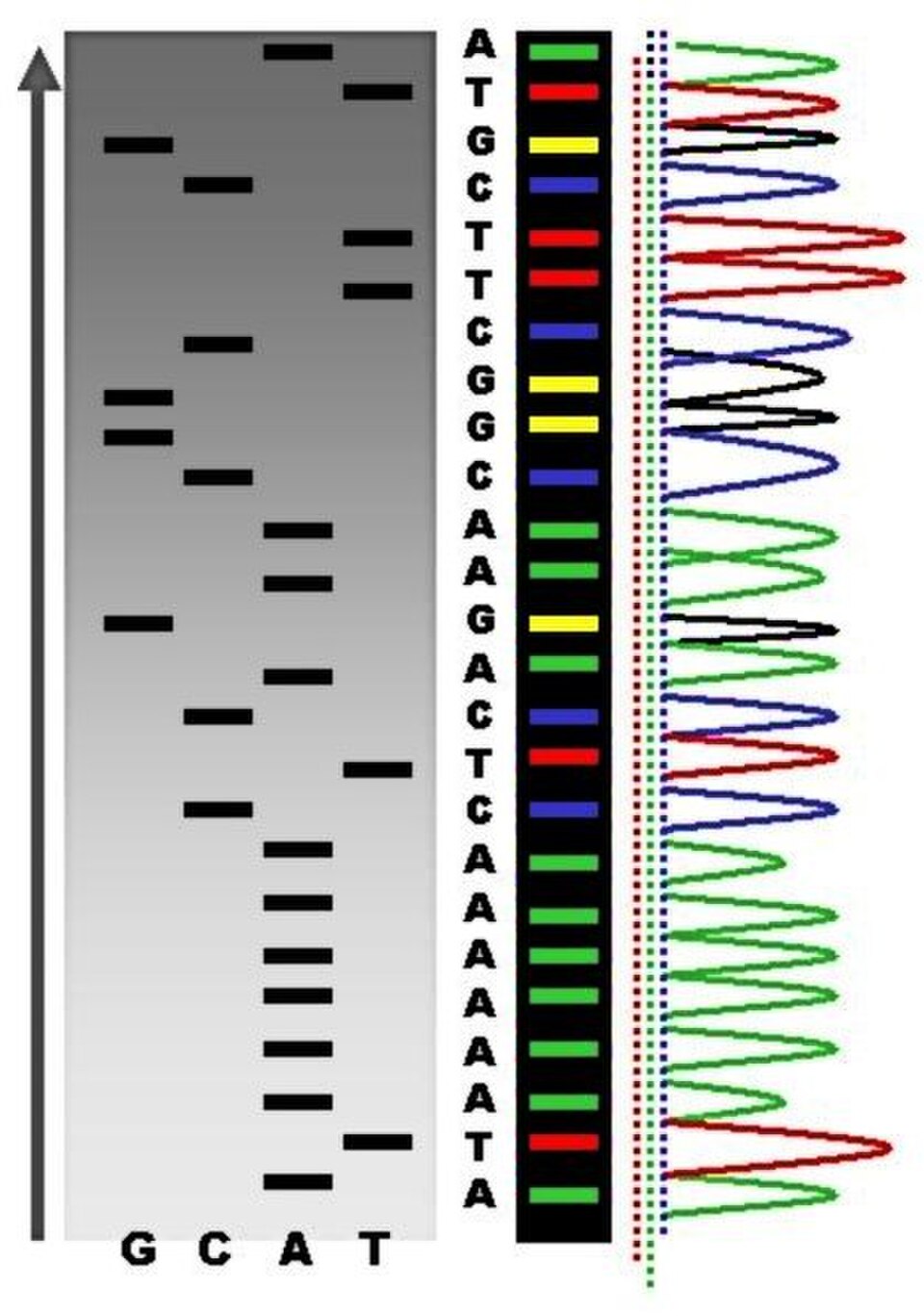
Métagénomique
Médecine
enquête médico-légale
Histoire
Découverte de la structure et de la fonction de l'ADN

Séquençage de l'ARN
Les premières méthodes de séquençage de l'ADN
Séquençage de génomes complets

Méthodes de séquençage à haut débit (HTS)

Méthodes de base
Séquençage de Maxam-Gilbert
Méthodes de terminaison de chaîne
Séquençage par synthèse
Séquençage par expansion
Séquençage à grande échelle et séquençage de novo
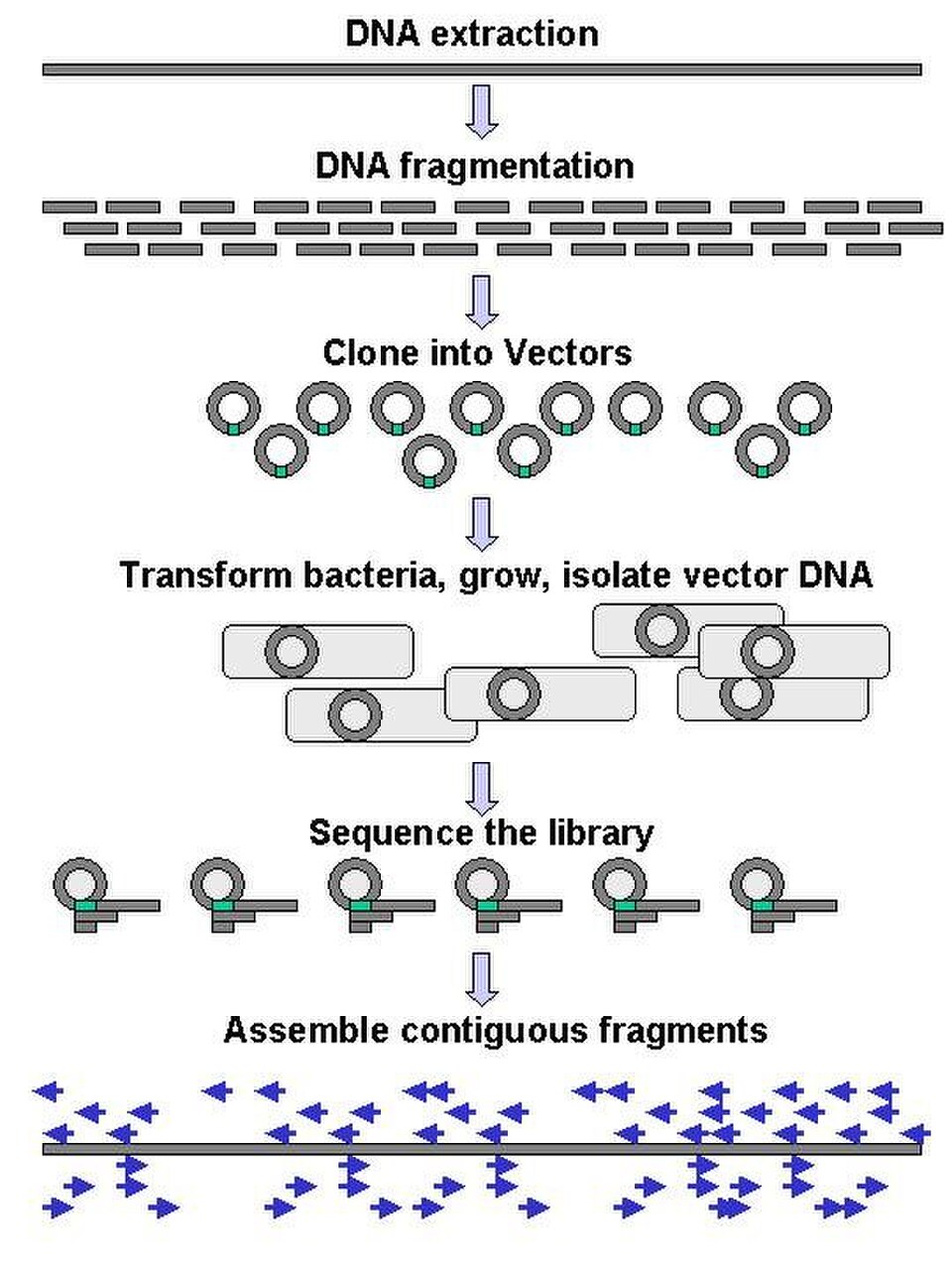
séquençage par fusil de chasse
Méthodes à haut débit
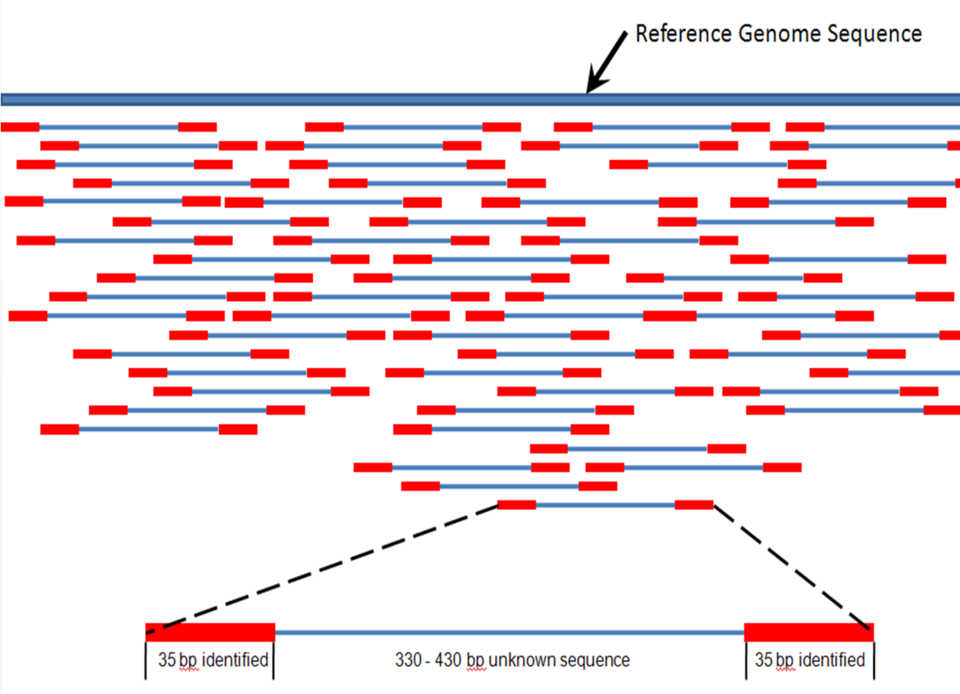
Méthode Longueur de lecture Précision (lecture unique, pas de consensus) Lectures par exécution Temps par course Coût par milliard de bases (en dollars américains) Avantages Inconvénients Séquençage en temps réel de molécules uniques (Pacific Biosciences) 30 000 pb ( N50 ) ; Précision de lecture brute de 87 % 4 000 000 par cellule SMRT Sequel 2, 100 à 200 gigabases 30 minutes à 20 heures 7,2 $ - 43,3 $ Rapide. Détecte 4 mC, 5 mC, 6 mA. Débit modéré. Le matériel peut être très coûteux. Semi-conducteur ionique (séquençage Ion Torrent) jusqu'à 600 pb 99,6 % jusqu'à 80 millions 2 heures 66,8 $ - 950 $ Équipement moins cher. Rapide. Erreurs d'homopolymères. Pyroséquençage (454) 700 pb 99,9% 1 million 24 heures 10 000 $ Format long. Rapide. Les tirages sont coûteux. Erreurs d'homopolymères. Séquençage par synthèse (Illumina) MiniSeq, NextSeq : 75–300 pb ; 99,9 % (Phred30) MiniSeq/MiSeq : 1 à 25 millions ; 1 à 11 jours, selon le séquenceur et la longueur de lecture spécifiée 5 $ à 150 $ Potentiel de rendement de séquençage élevé, selon le modèle de séquenceur et l'application souhaitée. Le matériel peut être très coûteux. Il nécessite de fortes concentrations d'ADN. Synthèse d'ancres de sondes combinatoires (cPAS-BGI/MGI) BGISEQ-50 : 35-50 pb ; 99,9 % (Phred30) BGISEQ-50 : 160 M ; 1 à 9 jours selon l'instrument, la longueur de lecture et le nombre de cellules de flux exécutées simultanément. 5 $ – 120 $ Séquençage par ligation (séquençage SOLiD) 50+35 ou 50+50 bp 99,9% 1,2 à 1,4 milliard 1 à 2 semaines 60 à 130 $ Faible coût par base. Plus lente que les autres méthodes. Présente des difficultés pour le séquençage des séquences palindromiques. Séquençage nanopore Dépendant de la préparation de la bibliothèque, et non du périphérique, l'utilisateur choisit donc la longueur de lecture (jusqu'à 2 272 580 pb signalés ). ~92–97% lecture unique en fonction de la longueur de lecture sélectionnée par l'utilisateur Données diffusées en temps réel. Choisissez une période de 1 min à 48 h. 7 à 100 $ Lectures individuelles les plus longues. Communauté d'utilisateurs accessible. Portable (format de paume). Débit inférieur à celui des autres machines, précision de lecture unique de l'ordre de 90 s. Séquençage GenapSys Environ 150 pb à extrémité unique 99,9 % (Phred30) 1 à 16 millions Environ 24 heures 667 $ Faible coût de l'instrument (10 000 $) Terminaison de chaîne (séquençage Sanger) 400 à 900 pb 99,9% N / A 20 minutes à 3 heures 2 400 000 $ Utile pour de nombreuses applications. Plus coûteuse et peu pratique pour les projets de séquençage de grande envergure, cette méthode nécessite également l'étape fastidieuse du clonage de plasmides ou de la PCR. Méthodes de séquençage à lecture longue
Méthodes de séquençage à lecture courtele séquençage de signatures massivement parallèle (MPSS, également appelé séquençage de nouvelle génération), a été développée dans les années 1990 chez Lynx Therapeutics, une société fondée en 1992 par Sydney Brenner et Sam Eletr . Le MPSS était une méthode basée sur des billes magnétiques qui utilisait une approche complexe de ligation d'adaptateurs suivie de leur décodage, la séquence étant lue par incréments de quatre nucléotides. Cette méthode la rendait sensible aux biais spécifiques à la séquence ou à la perte de séquences spécifiques. En raison de sa complexité, le MPSS était uniquement réalisé en interne par Lynx Therapeutics et aucun séquenceur d'ADN n'était vendu à des laboratoires indépendants. La fusion de Lynx Therapeutics avec Solexa (rachetée ultérieurement par Illumina ) en 2004 a conduit au développement du séquençage par synthèse, une approche plus simple acquise auprès de Manteia Predictive Medicine , qui a rendu le MPSS obsolète. Cependant, les propriétés essentielles des données de sortie MPSS étaient typiques des types de données à haut débit ultérieurs, notamment des centaines de milliers de courtes séquences d'ADN. Dans le cas de MPSS, celles-ci étaient généralement utilisées pour le séquençage d'ADNc afin de mesurer les niveaux d'expression génique .
Séquençage du polony
Séquençage Illumina (Solexa)

Synthèse d'ancres de sondes combinatoires (cPAS)

séquençage de semi-conducteurs Ion Torrent
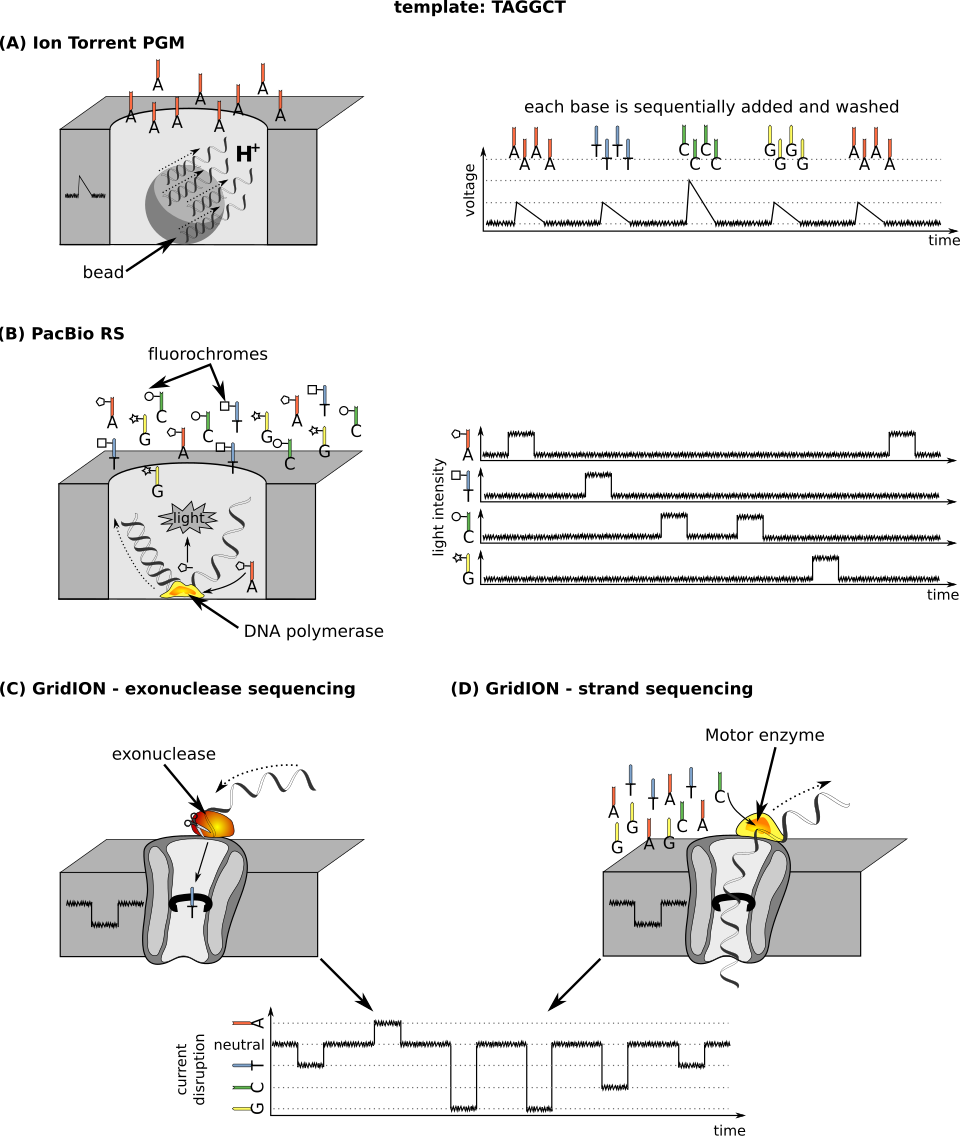
séquençage de nanobilles d'ADN
Séquençage de molécules uniques par hélioscope
Systèmes microfluidiques
Séquençage de l'ADN par courants tunnel
Séquençage par hybridation
Séquençage par spectrométrie de masse
Séquençage Sanger microfluidique
Séquençage à haut débit de virus in vitro
part de marché
Préparation des échantillons
Initiatives de développement

Défis informatiques
Lire la taille
Nom de l'algorithme Type d'algorithme Cutadapt Somme cumulée ConDeTri Windows FILTRE ERNE Somme cumulée Tondeuse de qualité FASTX Windows PRINSEQ Windows Trimmomatic Windows SolexaQA Windows SolexaQA-BWA Somme cumulée Faucille Windows Questions éthiques
Séquençage de l'ADN
Extrait d'une série sur Génétique Composants clés Chromosome ADN ARN Génome Hérédité Nucléotide Mutation variation génétique Allèle Acide aminé Contour Indice Histoire et sujets...
Médecine personnalisée