
En biologie moléculaire et en génétique , l'annotation de l'ADN ou annotation du génome est le processus de description de la structure et de la fonction des composants d'un génome , en les analysant et en les interprétant afin d'extraire leur signification biologique et de comprendre les processus biologiques auxquels ils participent. Entre autres choses, elle identifie l'emplacement des gènes et de toutes les régions codantes d'un génome et détermine ce que font ces gènes.
L'annotation est effectuée après le séquençage et l'assemblage d'un génome , et constitue une étape nécessaire de l'analyse du génome avant que la séquence ne soit déposée dans une base de données et décrite dans un article publié. Bien que la description des gènes individuels et de leurs produits ou fonctions soit suffisante pour considérer cette description comme une annotation, la profondeur de l'analyse rapportée dans la littérature pour différents génomes varie considérablement, certains rapports incluant des informations supplémentaires qui vont au-delà d'une simple annotation. De plus, en raison de la taille et de la complexité des génomes séquencés, l'annotation de l'ADN n'est pas effectuée manuellement, mais est plutôt automatisée par des moyens informatiques. Cependant, les conclusions tirées des résultats obtenus nécessitent une analyse manuelle par des experts.
L'annotation de l'ADN est classée en deux catégories : l'annotation structurelle , qui identifie et délimite les éléments d'un génome, et l'annotation fonctionnelle , qui attribue des fonctions à ces éléments. Ce n'est pas la seule façon dont elle a été catégorisée, car plusieurs alternatives, telles que les classifications basées sur les dimensions et les classifications basées sur les niveaux , ont également été proposées.
Histoire
La première génération d'annotateurs de génomes utilisait des méthodes ab initio locales , qui se basaient uniquement sur les informations pouvant être extraites de la séquence d'ADN à l'échelle locale, c'est-à-dire un cadre de lecture ouvert (ORF) à la fois. Elles sont apparues comme une nécessité pour gérer l'énorme quantité de données produites par les techniques de séquençage d'ADN Maxam-Gilbert et Sanger développées à la fin des années 1970. Le premier logiciel utilisé pour analyser les lectures de séquençage est le logiciel Staden Package , créé par Rodger Staden en 1977. Il effectuait plusieurs tâches liées à l'annotation, telles que le nombre de bases et de codons . En fait, l'utilisation des codons était la principale stratégie utilisée par plusieurs premières méthodes de prédiction de séquences codantes de protéines (CDS), basées sur l'hypothèse que les régions les plus traduites d'un génome contiennent des codons avec les ARNt correspondants les plus abondants (les molécules responsables du transport des acides aminés vers le ribosome pendant la synthèse des protéines) permettant une traduction plus efficace. On savait également que c’était le cas pour les codons synonymes , qui sont souvent présents dans les protéines exprimées à un niveau inférieur.
L'avènement des génomes complets dans les années 1990 (le premier étant le génome d' Haemophilus influenzae séquencé en 1995) a introduit une deuxième génération d'annotateurs. Tout comme dans la génération précédente, ils ont réalisé l'annotation par des méthodes ab initio , mais maintenant appliquées à l'échelle du génome. Les modèles de Markov sont la force motrice de nombreux algorithmes utilisés dans les annotateurs de cette génération ; ces modèles peuvent être considérés comme des graphes orientés où les nœuds représentent différents signaux génomiques (tels que les sites de démarrage de la transcription et de la traduction ) reliés par des flèches représentant le balayage de la séquence. Pour s'assurer qu'un modèle de Markov détecte un signal génomique, il doit d'abord être entraîné sur une série de signaux génomiques connus. Les résultats des modèles de Markov dans le contexte de l'annotation incluent les probabilités de chaque type d'élément génomique dans chaque partie du génome, et un modèle de Markov précis attribuera des probabilités élevées aux annotations correctes et des probabilités faibles aux annotations incorrectes.
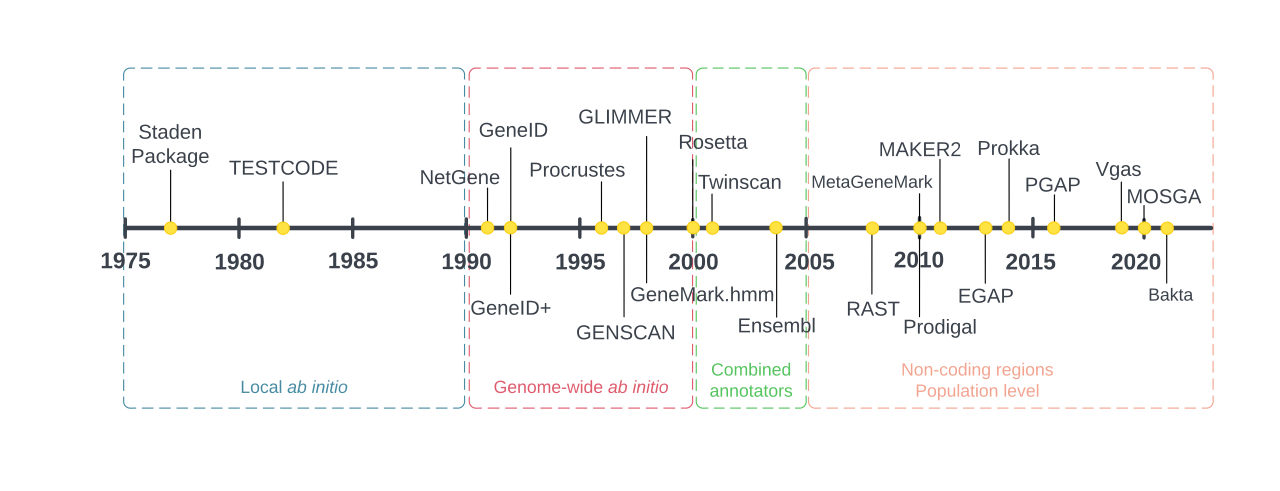
Au début et au milieu des années 2000, de plus en plus de génomes séquencés ont commencé à être disponibles, ainsi que de nombreuses séquences protéiques obtenues expérimentalement. Les annotateurs de génomes ont donc commencé à utiliser des méthodes basées sur l'homologie, lançant ainsi la troisième génération d'annotation du génome. Ces nouvelles méthodes ont permis aux annotateurs non seulement de déduire des éléments génomiques par des moyens statistiques (comme dans les générations précédentes), mais aussi d'effectuer leur tâche en comparant la séquence annotée avec d'autres séquences déjà existantes et validées. Ces annotateurs dits combinateurs, qui effectuent à la fois des annotations ab initio et basées sur l'homologie, nécessitent des algorithmes d'alignement rapides pour identifier les régions d' homologie .
À la fin des années 2000, l'annotation du génome s'est orientée vers l'identification des régions non codantes de l'ADN, ce qui a été rendu possible grâce à l'apparition de méthodes d'analyse des sites de liaison des facteurs de transcription , des sites de méthylation de l'ADN , de la structure de la chromatine et d'autres techniques d'analyse de l'ARN et des régions régulatrices . D'autres annotateurs du génome ont également commencé à se concentrer sur les études au niveau de la population représentée par le pangénome ; ce faisant, par exemple, les pipelines d'annotation garantissent que les gènes centraux d'un clade se trouvent également dans de nouveaux génomes du même clade. Les deux stratégies d'annotation constituent la quatrième génération d'annotateurs du génome.
Dans les années 2010, les séquences génomiques de plus d'un millier d'individus humains (grâce au projet 1000 génomes ) et de plusieurs organismes modèles sont devenues disponibles. L'annotation du génome reste donc un défi majeur pour les scientifiques qui étudient le génome humain et d'autres génomes.
Annotation structurelle
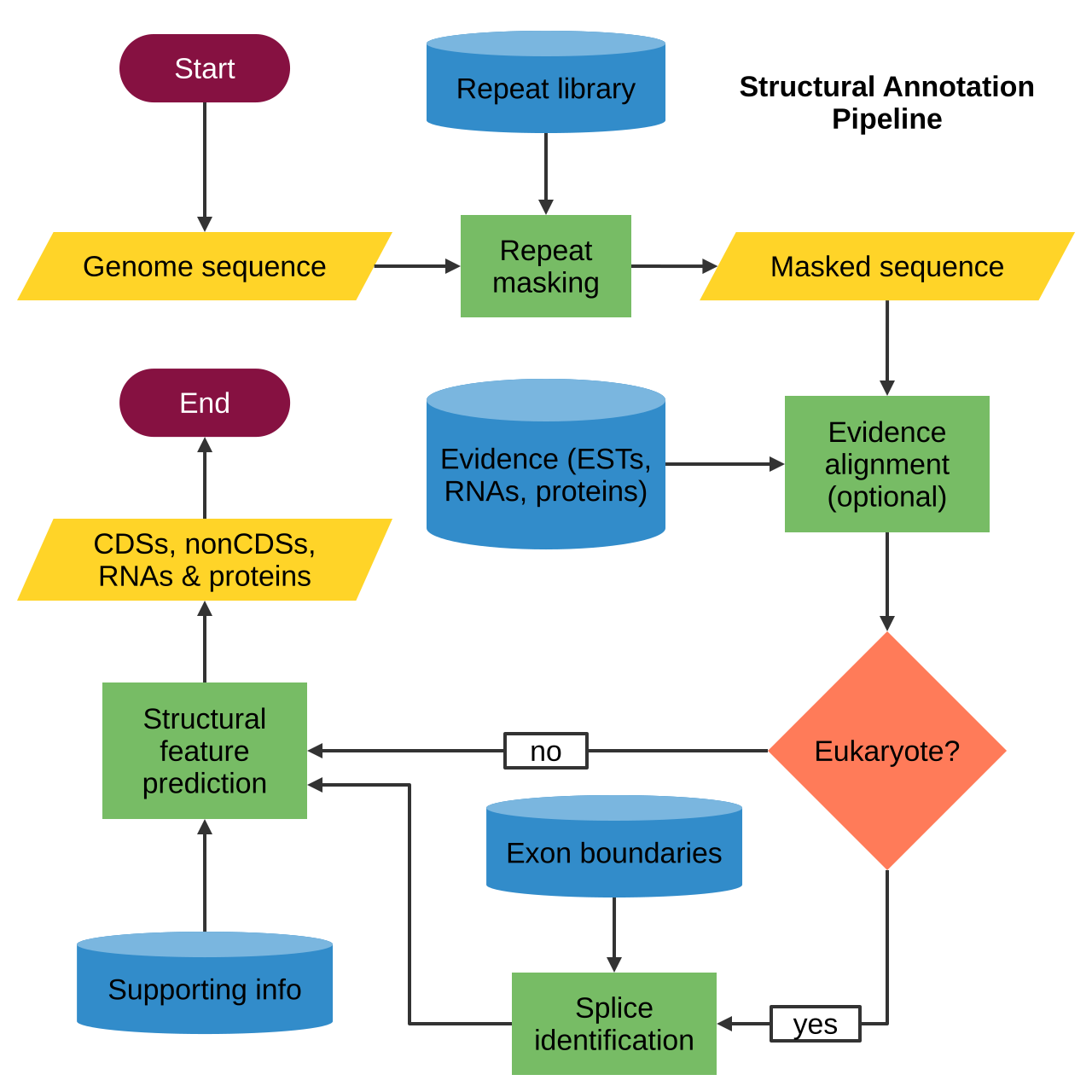
L'annotation structurelle décrit l'emplacement précis des différents éléments d'un génome, tels que les cadres de lecture ouverts (ORF), les séquences codantes (CDS), les exons , les introns , les répétitions , les sites d'épissage , les motifs régulateurs , les codons de départ et d'arrêt et les promoteurs . Les principales étapes de l'annotation structurelle sont les suivantes :
- Répéter l'identification et le masquage.
- Alignement des preuves (facultatif).
- Identification de l'épissure (uniquement chez les eucaryotes).
- Prédiction de caractéristiques (séquences codantes et non codantes).
Répéter l'identification et le masquage
La première étape de l'annotation structurelle consiste à identifier et à masquer les répétitions , qui comprennent des séquences de faible complexité (telles que AGAGAGAG, ou des segments monopolymériques comme TTTTTTTTT), et des transposons (qui sont des éléments plus grands avec plusieurs copies à travers le génome). Les répétitions sont un composant majeur des génomes procaryotes et eucaryotes ; par exemple, entre 0 % et plus de 42 % des génomes procaryotes sont constitués de répétitions et les trois quarts du génome humain sont composés d'éléments répétitifs.
L'identification des répétitions est difficile pour deux raisons principales : elles sont mal conservées et leurs limites ne sont pas clairement définies. Pour cette raison, des bibliothèques de répétitions doivent être créées pour le génome d'intérêt, ce qui peut être réalisé avec l'une des méthodes suivantes :
- Méthodes de novo . Les répétitions sont identifiées en détectant et en regroupant des paires de séquences à différents endroits dont la similarité est supérieure à un seuil minimum de conservation de séquence dans une comparaison d'auto-génome, ne nécessitant ainsi aucune information préalable sur la structure ou les séquences répétées. L'inconvénient de ces méthodes est qu'elles peuvent identifier n'importe quelle séquence répétée, pas seulement les transposons, et peuvent inclure des séquences codantes conservées (CDS), ce qui fait d'un post-traitement minutieux une étape indispensable pour supprimer ces séquences. Elle peut également exclure des régions apparentées qui se sont dégradées au fil du temps et peut regrouper des éléments qui n'ont aucun lien dans leur histoire évolutive.
- Méthodes basées sur l'homologie . Les répétitions sont identifiées par la similarité ( homologie ) des répétitions connues stockées dans une base de données organisée. Ces méthodes sont plus susceptibles de trouver de vrais transposons, même en plus faible quantité, par rapport aux méthodes de novo , mais sont biaisées en faveur des familles précédemment identifiées.
- Méthodes basées sur la structure . Les répétitions sont identifiées sur la base de modèles de leur structure, plutôt que de répétitions ou de similarités. Elles sont capables d'identifier de vrais transposons (tout comme celles basées sur l'homologie), mais ne sont pas biaisées par des éléments connus. Cependant, elles sont très spécifiques à chaque classe de répétitions et, en tant que telles, sont moins universellement applicables.
- Méthodes génomiques comparatives . Les répétitions sont identifiées comme des perturbations d'une ou plusieurs séquences dans un alignement de séquences multiples produites par de grandes régions d'insertion . Bien que cette stratégie évite le problème de limites mal définies qui existe dans d'autres méthodes, elle dépend fortement de la qualité de l'assemblage et du niveau d'activité des transposons dans les génomes en question.
Une fois les régions répétitives d'un génome identifiées, elles sont masquées. Le masquage consiste à remplacer les lettres des nucléotides (A, C, G ou T) par d'autres lettres. Ce faisant, ces régions seront marquées comme répétitives et les analyses en aval les traiteront en conséquence. Les régions répétitives peuvent produire des problèmes de performances si elles ne sont pas masquées, et peuvent même produire de fausses preuves pour l'annotation des gènes (par exemple, traiter un cadre de lecture ouvert (ORF) dans un transposon comme un exon ) Selon les lettres utilisées pour le remplacement, le masquage peut être classé comme doux ou dur : dans le masquage doux , les régions répétitives sont indiquées par des lettres minuscules (a, c, g ou t), tandis que dans le masquage dur , les lettres de ces régions sont remplacées par des N. De cette façon, par exemple, le masquage doux peut être utilisé pour exclure les correspondances de mots et éviter d'initier un alignement dans ces régions, et le masquage dur, en dehors de tout cela, peut également exclure les régions masquées des scores d'alignement.
Alignement des preuves
L'étape suivante après le masquage du génome consiste généralement à aligner toutes les preuves de transcription et de protéines disponibles avec le génome analysé, c'est-à-dire à aligner toutes les étiquettes de séquence exprimées (EST), les ARN et les protéines connus de l'organisme annoté avec le génome. Bien que cela soit facultatif, cela peut améliorer l'élucidation de la séquence génétique car les ARN et les protéines sont des produits directs de séquences codantes.
Si des données de séquençage de l'ARN sont disponibles, elles peuvent être utilisées pour annoter et quantifier tous les gènes et leurs isoformes situés dans le génome correspondant, en fournissant non seulement leurs emplacements, mais aussi leurs taux d'expression. Cependant, les transcriptions ne fournissent pas suffisamment d'informations pour la prédiction des gènes, car elles peuvent être impossibles à obtenir à partir de certains gènes, elles peuvent coder des opérones de plus d'un gène et leurs codons de départ et d'arrêt ne peuvent pas être déterminés en raison de décalages de cadre et de facteurs d'initiation de la traduction . Pour résoudre ce problème, des approches basées sur la protéogénomique sont utilisées, qui utilisent des informations provenant de protéines exprimées souvent dérivées de la spectrométrie de masse .
Identification des épissures
L'annotation des génomes eucaryotes présente une difficulté supplémentaire en raison de l'épissage de l'ARN , un processus post-transcriptionnel dans lequel les introns (régions non codantes) sont supprimés et les exons (régions codantes) sont joints. les séquences codantes eucaryotes (CDS) sont discontinues et, pour assurer leur identification correcte, les régions introniques doivent être filtrées. Pour ce faire, les pipelines d'annotation doivent trouver les limites exon-intron, et plusieurs méthodologies ont été développées à cet effet. Une solution consiste à utiliser les limites d'exons connues pour l'alignement ; par exemple, de nombreux introns commencent par GT et se terminent par AG. Cette approche, cependant, ne peut pas détecter de nouvelles limites, il existe donc des alternatives comme les algorithmes d'apprentissage automatique qui sont formés sur des limites d'exons connues et des informations de qualité pour en prédire de nouvelles. Les prédicteurs de nouvelles limites d'exons nécessitent généralement des algorithmes efficaces de compression et d'alignement des données, mais ils sont sujets à des échecs dans les limites situées dans des régions avec une faible couverture de séquence ou des taux d'erreur élevés produits pendant le séquençage.
Prédiction des fonctionnalités
Un génome est divisé en régions codantes et non codantes , et la dernière étape de l'annotation structurelle consiste à identifier ces caractéristiques au sein du génome. En fait, la tâche principale de l'annotation du génome est la prédiction des gènes , c'est pourquoi de nombreuses méthodes ont été développées à cette fin. La prédiction des gènes est un terme trompeur, car la plupart des prédicteurs de gènes n'identifient que les séquences codantes (CDS) et ne signalent pas les régions non traduites (UTR) ; pour cette raison, la prédiction CDS a été proposée comme un terme plus précis. Les prédicteurs CDS détectent les caractéristiques du génome grâce à des méthodes appelées capteurs , qui incluent des capteurs de signal qui identifient les signaux de sites fonctionnels tels que les promoteurs et les sites polyA , et des capteurs de contenu qui classent les séquences d'ADN en contenu codant et non codant. Alors que les prédicteurs CDS procaryotes traitent principalement des cadres de lecture ouverts (ORF), qui sont des segments d'ADN entre les codons de départ et d'arrêt , les prédicteurs CDS eucaryotes sont confrontés à un problème plus difficile en raison de l'organisation complexe des gènes eucaryotes. Les méthodes de prédiction CDS peuvent être classées en trois grandes catégories :
- Méthodes ab initio (également appelées statistiques, intrinsèques ou de novo). La prédiction CDS est basée uniquement sur les informations qui peuvent être extraites de la séquence d'ADN. Elles s'appuient sur des méthodes statistiques telles que le modèle de Markov caché (HMM). Certaines méthodes utilisent deux génomes ou plus pour déduire les taux et les modèles de mutation locaux le long du génome.
- Méthodes basées sur l'homologie (également appelées empiriques, fondées sur des preuves ou extrinsèques). La prédiction CDS est basée sur la similarité avec des séquences connues. Plus précisément, elle effectue des alignements de la séquence analysée avec des étiquettes de séquence exprimée (EST), de l'ADN complémentaire (ADNc) ou des séquences protéiques .
- Combineurs . La prédiction CDS est réalisée par une combinaison des deux méthodes mentionnées ci-dessus.
Annotation fonctionnelle
L'annotation fonctionnelle attribue des fonctions aux éléments génomiques trouvés par annotation structurelle, en les reliant à des processus biologiques tels que le cycle cellulaire , la mort cellulaire , le développement , le métabolisme , etc. Elle peut également être utilisée comme un contrôle de qualité supplémentaire en identifiant les éléments qui peuvent avoir été annotés par erreur.
Prédiction de la fonction de séquence de codage

L'annotation fonctionnelle des gènes nécessite un vocabulaire contrôlé (ou ontologie) pour nommer les caractéristiques fonctionnelles prédites. Cependant, comme il existe de nombreuses façons de définir les fonctions des gènes, le processus d'annotation peut être entravé lorsqu'il est effectué par différents groupes de recherche. Il est donc nécessaire d'utiliser un vocabulaire contrôlé standardisé, dont le plus complet est l' ontologie des gènes (GO). Elle classe les propriétés fonctionnelles dans l'une des trois catégories (fonction moléculaire, processus biologique et composant cellulaire) et les organise dans un graphe acyclique dirigé , dans lequel chaque nœud est une fonction particulière, et chaque arête (ou flèche) entre deux nœuds indique une relation parent-enfant ou sous-catégorie-catégorie. En 2020, GO est le vocabulaire contrôlé le plus utilisé pour l'annotation fonctionnelle des gènes, suivi du catalogue fonctionnel MIPS (FunCat).
Certaines méthodes conventionnelles d'annotation fonctionnelle sont basées sur l'homologie , qui s'appuient sur des outils de recherche d'alignement local . Son principe est que la conservation élevée des séquences entre deux éléments génomiques implique que leur fonction est également conservée. Les paires de séquences homologues qui sont apparues par paralogie , orthologie ou xénologie remplissent généralement une fonction similaire. Cependant, les séquences orthologues doivent être traitées avec prudence pour deux raisons : (1) elles peuvent avoir des noms différents selon le moment où elles ont été annotées à l'origine, et (2) elles peuvent ne pas remplir le même rôle fonctionnel dans deux organismes différents. Les annotateurs font souvent référence à une séquence analogue lorsqu'aucune paralogie, orthologie ou xénologie n'a été trouvée. Les méthodes basées sur l'homologie présentent plusieurs inconvénients, tels que des erreurs dans la base de données, une faible sensibilité/spécificité, l'incapacité à faire la distinction entre paralogie et homologie, des scores artificiellement élevés en raison de la présence de régions de faible complexité et une variation significative au sein d'une famille de protéines.
L'annotation fonctionnelle peut être réalisée par des méthodes probabilistes. La distribution des acides aminés hydrophiles et hydrophobes indique si une protéine est située dans une solution ou une membrane. Des motifs de séquence spécifiques fournissent des informations sur les modifications post-traductionnelles et l'emplacement final de toute protéine donnée. Les méthodes probabilistes peuvent être associées à un vocabulaire contrôlé, tel que GO ; par exemple, les réseaux d' interaction protéine-protéine (PPI) placent généralement les protéines ayant des fonctions similaires à proximité les unes des autres.
Les méthodes d'apprentissage automatique sont également utilisées pour générer des annotations fonctionnelles pour de nouvelles protéines basées sur des termes GO. En général, elles consistent à construire un classificateur binaire pour chaque terme GO, qui sont ensuite joints pour faire des prédictions sur des termes GO individuels (formant un classificateur multiclasse ) pour lesquels des scores de confiance sont ensuite obtenus. La machine à vecteurs de support (SVM) est le classificateur binaire le plus largement utilisé dans l'annotation fonctionnelle ; cependant, d'autres algorithmes, tels que les k-plus proches voisins (kNN) et le réseau neuronal convolutionnel (CNN), ont également été utilisés.
Les méthodes de classification binaire ou multiclasse pour l'annotation fonctionnelle produisent généralement des résultats moins précis car elles ne prennent pas en compte les interrelations entre les termes GO. Les méthodes plus avancées qui prennent en compte ces interrelations le font soit par une approche plate, soit par une approche hiérarchique, qui se distinguent par le fait que la première ne prend pas en compte la structure de l'ontologie, tandis que la seconde le fait. Certaines de ces méthodes compressent les termes GO par factorisation matricielle ou par hachage , augmentant ainsi leurs performances.
Prédiction de la fonction de séquence non codante
Les séquences non codantes (ADNnc) sont celles qui ne codent pas pour les protéines. Elles comprennent des éléments tels que les pseudogènes, les duplications segmentaires, les sites de liaison et les gènes d'ARN.
Les pseudogènes sont des copies mutées de gènes codant pour des protéines qui ont perdu leur fonction de codage en raison d'une perturbation de leur cadre de lecture ouvert (ORF), les rendant intraduisibles . Ils peuvent être identifiés à l'aide de l'une des deux méthodes suivantes :
- Méthode basée sur l'homologie . Les pseudogènes sont identifiés en recherchant des séquences similaires à des gènes fonctionnels mais contenant des mutations qui produisent une perturbation dans leur ORF. Cette méthode ne permet pas de déterminer la relation évolutive entre un pseudogène et son gène parent ni le temps écoulé depuis que l'événement s'est produit.
- Méthode basée sur la phylogénie . Les pseudogènes sont identifiés au moyen d'une analyse phylogénétique. Tout d'abord, un arbre d'espèces de l'espèce d'intérêt et un arbre phylogénétique du gène (ou de la famille de gènes) d'intérêt sont construits. Les deux sont ensuite comparés pour identifier une espèce qui a perdu le gène. Ensuite, dans le génome de l'espèce où le gène n'a pas été trouvé, une séquence orthologue au gène identifié dans l'espèce la plus proche est recherchée. Enfin, si cette séquence orthologue présente une disruption dans son ORF (et qu'elle répond à d'autres critères, tels que l'analyse des données RNA-Seq , le rapport dN/dS , etc.), cela signifie que la séquence est bien un pseudogène.
Les duplications segmentaires sont des segments d'ADN de plus de 1000 paires de bases qui sont répétés dans le génome avec plus de 90 % d'identité de séquence. Deux stratégies utilisées pour leur identification sont WGAC et WSSD :
- Comparaison de l'assemblage du génome entier (WGAC). Elle aligne l'ensemble du génome sur lui-même afin d'identifier les séquences répétées après avoir filtré les répétitions communes ; elle ne nécessite pas que les lectures originales soient utilisées pour l'assemblage.
- Détection de séquences Shotgun sur génome entier (WSSD). Elle aligne les lectures originales avec le génome assemblé et recherche les régions avec une profondeur de lecture supérieure à la moyenne, qui sont généralement des signaux de duplication. Les duplications segmentaires identifiées par cette méthode mais pas par WGAC sont probablement des duplications effondrées, ce qui signifie qu'elles ont été alignées par erreur sur la même région.
Les sites de liaison à l'ADN sont des régions de la séquence du génome qui se lient à des protéines spécifiques et interagissent avec elles. Ils jouent un rôle important dans la réplication et la réparation de l'ADN , la régulation transcriptionnelle et l'infection virale . La prédiction du site de liaison implique l'utilisation de l'une des deux méthodes suivantes :
- Méthodes basées sur la similarité des séquences . Elles consistent à identifier des séquences homologues dont les sites de liaison à l'ADN sont connus, ou à les aligner avec des protéines recherchées. Leurs performances sont généralement faibles car les séquences de liaison à l'ADN sont moins conservées .
- Méthodes basées sur la structure . Elles utilisent les informations structurelles tridimensionnelles des protéines pour prédire l'emplacement des sites de liaison à l'ADN.
L'ARN non codant (ARNnc), produit par les gènes d'ARN, est un type d'ARN qui n'est pas traduit en protéine. Il comprend des molécules telles que l'ARNt , l'ARNr , l'ARNsno et le microARN , ainsi que des transcrits de type ARNm non codants . La prédiction ab initio des gènes d'ARN dans un seul génome donne souvent des résultats inexacts (à l'exception de l'ARNmi), c'est pourquoi des méthodes comparatives multigénomes sont utilisées à la place. Ces méthodes s'intéressent spécifiquement aux structures secondaires de l'ARNnc, car elles sont conservées dans les espèces apparentées même lorsque leur séquence ne l'est pas. Par conséquent, en effectuant un alignement de séquences multiples, des informations plus utiles peuvent être obtenues pour leur prédiction. La recherche d'homologie peut également être utilisée pour identifier les gènes d'ARN, mais cette procédure est compliquée, en particulier chez les eucaryotes, en raison de la présence d'un grand nombre de répétitions et de pseudogènes.
Visualisation
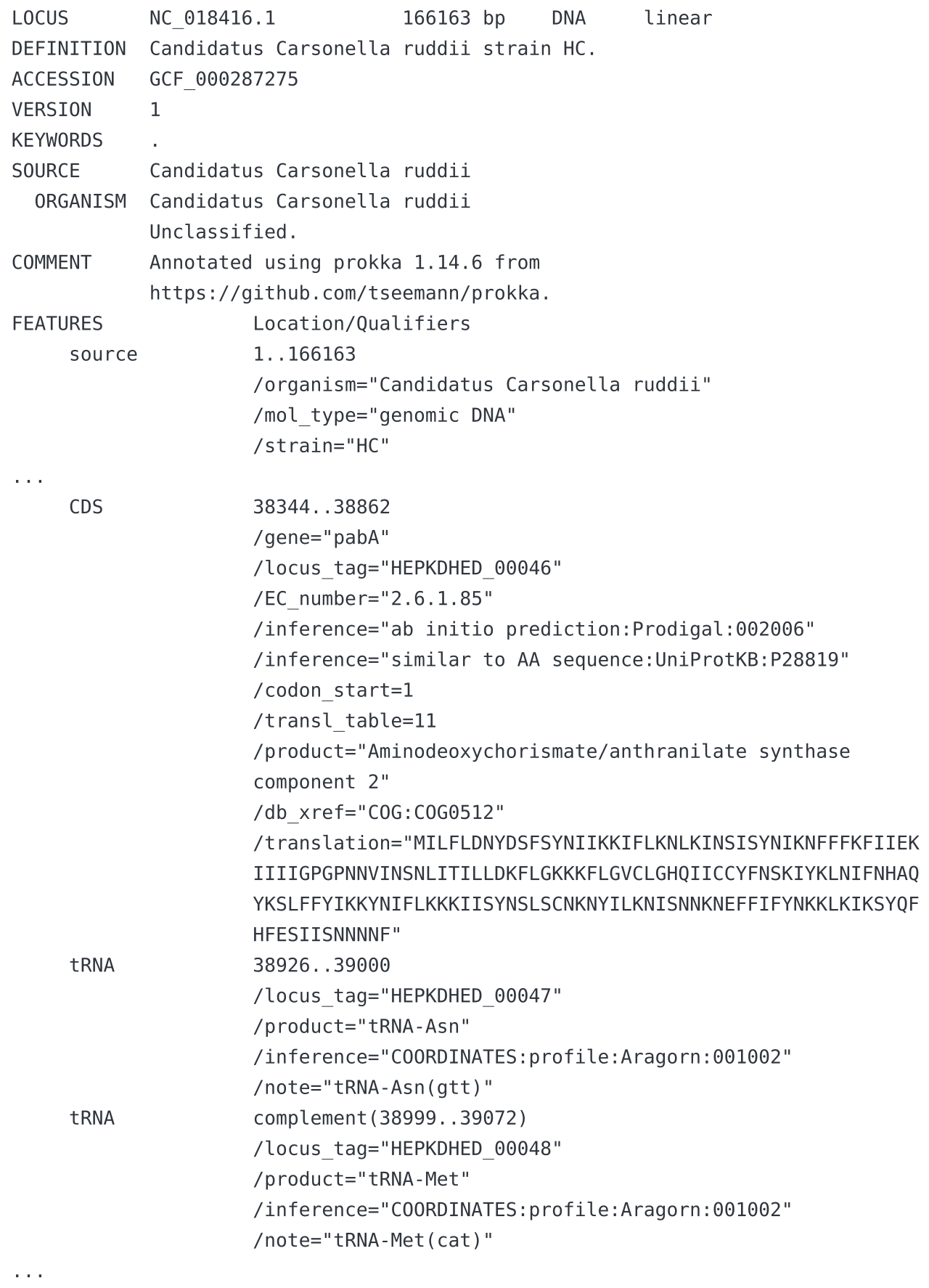
Formats de fichiers
La visualisation des annotations dans un navigateur de génome nécessite un fichier de sortie descriptif, qui doit décrire les structures intron - exon de chaque annotation, leurs codons de départ et d'arrêt , les UTR et les transcriptions alternatives, et doit idéalement inclure des informations sur les alignements de séquences et les prédictions génétiques qui prennent en charge chaque modèle de gène. Certains formats couramment utilisés pour décrire les annotations sont GenBank, GFF3 , GTF, BED et EMBL. Certains de ces formats utilisent des vocabulaires contrôlés et des ontologies pour définir leurs terminologies descriptives et garantir l'interopérabilité entre les outils d'analyse et de visualisation.
Navigateurs de génomes
Les navigateurs génomiques sont des produits logiciels qui simplifient l'analyse et la visualisation de grandes séquences génomiques et de données d'annotation pour obtenir des informations biologiques, via une interface graphique.
Les navigateurs génomiques peuvent être divisés en navigateurs génomiques basés sur le Web et navigateurs génomiques autonomes . Les premiers utilisent des informations provenant de bases de données et peuvent être classés en navigateurs multi-espèces (intégrant la séquence et les annotations de plusieurs organismes et favorisant l'analyse comparative entre espèces) et navigateurs spécifiques à une espèce (se concentrant sur un organisme et les annotations pour une espèce particulière). Ces derniers ne sont pas nécessairement liés à une base de données génomique spécifique, mais sont des navigateurs à usage général qui peuvent être téléchargés et installés en tant qu'application sur un ordinateur local.
Visualisation comparative des génomes
La génomique comparative vise à identifier les similitudes et les différences dans les caractéristiques génomiques, ainsi qu'à examiner les relations évolutives entre les organismes. Les outils de visualisation capables d'illustrer le comportement comparatif entre deux ou plusieurs génomes sont essentiels pour cette approche et peuvent être classés en trois catégories en fonction de la représentation des relations entre les génomes comparés :
- Dot Plots : Ce schéma permet uniquement de montrer l'alignement de deux génomes, un génome est représenté le long de l'axe horizontal et l'autre le long de l'axe vertical et les points du graphique représentent les éléments génomiques qui sont similaires entre ces deux annotations.
- Représentation linéaire : cette représentation utilise plusieurs pistes linéaires pour représenter plusieurs génomes et leurs caractéristiques, où « piste » est un concept qui fait référence à un type spécifique de caractéristique génomique à un emplacement génomique.
- Représentation circulaire : cette représentation facilite la comparaison de génomes microbiens ou viraux entiers. Dans ce mode de visualisation, des cercles et des arcs concentriques sont utilisés pour représenter des sections génomiques.
Contrôle de qualité
La qualité de l' assemblage de séquences influence la qualité de l'annotation, il est donc important d'évaluer la qualité de l'assemblage avant d'effectuer les étapes d'annotation suivantes. Afin de quantifier la qualité d'une annotation du génome, trois mesures ont été utilisées : le rappel , la précision et l'exactitude ; bien que ces mesures ne soient pas explicitement utilisées dans les projets d'annotation, mais plutôt dans les discussions sur la précision de la prédiction.
Les approches d'annotation communautaire sont d'excellentes techniques de contrôle de la qualité et de normalisation dans l'annotation du génome. Un rassemblement d'annotations qui a eu lieu en 2002 a conduit à la création des normes d'annotation utilisées par le projet d'analyse des humains et des vertébrés (HAVANA) de l'Institut Sanger.
Réannotation
Les projets d'annotation s'appuient souvent sur des annotations antérieures du génome d'un organisme. Cependant, ces annotations plus anciennes peuvent contenir des erreurs qui peuvent se propager aux nouvelles annotations. À mesure que de nouvelles technologies d'analyse du génome sont développées et que des bases de données plus riches deviennent disponibles, l'annotation de certains génomes plus anciens peut être mise à jour. Ce processus, connu sous le nom de réannotation, peut fournir aux utilisateurs de nouvelles informations sur le génome, notamment des détails sur les gènes et les fonctions des protéines. La réannotation est donc une approche utile dans le contrôle de la qualité.
Annotation de la communauté
L'annotation communautaire consiste à impliquer une communauté (scientifique ou non) dans des projets d'annotation du génome. Elle peut être classée dans les six catégories suivantes :
- Modèle d'usine : L'annotation est effectuée par un pipeline entièrement automatisé.
- Modèle de musée : une conservation manuelle par des experts est nécessaire pour interpréter les résultats d'un projet d'annotation.
- Modèle d'artisanat : l'annotation est décentralisée et est le résultat des efforts de différents conservateurs à temps partiel.
- Modèle de fête ou de jamboree : il consiste en un atelier intensif de courte durée avec des conservateurs de premier plan de la communauté. Il a été utilisé pour la première fois dans le projet d'annotation du génome de Drosophila melanogaster
- Annotateur béni : une variante du modèle de musée, appliquée dans le projet Knockout Mouse (KOMP), dans laquelle les conservateurs passent par une période de formation avant l'annotation, et ont ensuite accès à des outils d'annotation pour continuer leur travail.
- Approche du gardien : il s'agit d'une combinaison des modèles de jamboree et de l'industrie artisanale. Elle commence par un atelier d'annotation, suivi d'une collaboration décentralisée pour étendre et affiner l'annotation initiale. Elle a été utilisée pour les données de plusieurs espèces.
Une annotation communautaire est dite supervisée lorsqu'il y a un coordinateur qui gère le projet en demandant l'annotation d'éléments spécifiques à un nombre restreint d'experts. En revanche, lorsque n'importe qui peut entrer dans un projet et que la coordination est réalisée de manière décentralisée, on parle d'annotation communautaire non supervisée . L'annotation communautaire supervisée est de courte durée et limitée à la durée de l'événement, alors que la contrepartie non supervisée n'a pas cette limitation. Cependant, cette dernière a eu moins de succès que la première, probablement en raison d'un manque de temps, de motivation, d'incitation et/ou de communication.
Wikipédia a plusieurs projets Wikipédia visant à améliorer l'annotation. Le projet Wikipédia Gene , par exemple, exploite un robot qui collecte des données génétiques à partir de bases de données de recherche et crée des souches de gènes sur cette base. Le projet Wikipédia RNA cherche à rédiger des articles qui décrivent les ARN individuels et les familles d'ARN de manière accessible.
Applications
Diagnostic de la maladie
L'ontologie génétique est utilisée par les chercheurs pour établir une relation maladie-gène, car GO aide à l'identification de nouveaux gènes, aux altérations de leur expression, de leur distribution et de leur fonction dans un ensemble différent de conditions, comme la maladie par rapport à la santé. Des bases de données de ces relations maladie-gène de différents organismes ont été créées, telles que Plant-Pathogen Ontology, Plant-Associated Microbe Gene Ontology ou DisGeNET. Et d'autres ont été implémentées dans des bases de données préexistantes comme Rat Disease Ontology dans la base de données Rat Genome.
Bioremédiation
Une grande diversité d' enzymes cataboliques impliquées dans la dégradation des hydrocarbures par certaines souches bactériennes sont codées par des gènes localisés dans leurs éléments génétiques mobiles (MGEs). L'étude de ces éléments est d'une grande importance dans le domaine de la bioremédiation, puisque récemment l'inoculation de souches sauvages ou génétiquement modifiées avec ces MGEs a été recherchée afin d'acquérir ces capacités de dégradation des hydrocarbures. En 2013, Phale et al. ont publié l'annotation du génome d'une souche de Pseudomonas putida (CSV86), une bactérie connue pour sa préférence pour le naphtalène et d'autres composés aromatiques par rapport au glucose comme source de carbone et d'énergie. Afin de trouver les MGEs de cette bactérie, son génome a été annoté à l'aide de RAST et du NCBI Prokaryotic Genome Annotation Pipeline (PGAP), et l'identification de neuf éléments mobiles a été possible avec la base de données Insertion Sequence (IS) Finder. Cette analyse a conclu à la localisation des gènes de la voie supérieure de dégradation du naphtalène, juste à côté des gènes codant l'ARNt-Gly et l'intégrase, ainsi qu'à l'identification des gènes codant les enzymes impliquées dans la dégradation du salicylate , du benzoate , du 4-hydroxybenzoate , de l'acide phénylacétique , de l'acide hydroxyphénylacétique, et à la reconnaissance d'un opéron impliqué dans le transport du glucose dans la souche.
L'analyse de l'ontologie génétique est d'une grande importance dans l'annotation fonctionnelle, et en particulier dans la bioremédiation, elle peut être appliquée pour connaître les relations entre les gènes de certains micro-organismes avec leurs fonctions et leur rôle dans la remédiation de certains contaminants. C'est l'approche de l'investigation et de l'identification de la souche Halomonas zincidurans B6(T), une bactérie avec trente et un gènes codant pour la résistance aux métaux lourds , en particulier au zinc et Stenotrophomonas sp. DDT-1, une souche capable d'utiliser le DDT comme seule source de carbone et d'énergie, pour ne citer que quelques exemples.
Logiciel
Les gènes d'un génome eucaryote peuvent être annotés à l'aide de divers outils d'annotation tels que FINDER. Un pipeline d'annotation moderne peut prendre en charge une interface Web conviviale et une conteneurisation logicielle telle que MOSGA. Les pipelines d'annotation modernes pour les génomes procaryotes sont Bakta, Prokka et PGAP.
Le Centre national d'ontologie biomédicale développe des outils d'annotation automatisée des enregistrements de bases de données en fonction des descriptions textuelles de ces enregistrements.
En tant que méthode générale, dcGO dispose d'une procédure automatisée permettant de déduire statistiquement les associations entre les termes d'ontologie et les domaines protéiques ou les combinaisons de domaines à partir des annotations existantes au niveau des gènes/protéines.
Une variété d’outils logiciels ont été développés pour permettre aux scientifiques de visualiser et de partager les annotations du génome, tels que MAKER.
L'annotation du génome est un domaine de recherche actif et implique un certain nombre d'organisations différentes de la communauté des sciences de la vie qui publient les résultats de leurs efforts dans des bases de données biologiques accessibles au public via le Web et d'autres moyens électroniques. Voici une liste alphabétique des projets en cours concernant l'annotation du génome :