Le nucléoïde (qui signifie en forme de noyau ) est une région de forme irrégulière à l'intérieur de la cellule procaryote qui contient tout ou la majeure partie du matériel génétique . Le chromosome d'un procaryote typique est circulaire et sa longueur est très grande par rapport aux dimensions de la cellule, il doit donc être compacté pour s'adapter. Contrairement au noyau d'une cellule eucaryote , il n'est pas entouré d'une membrane nucléaire . Au lieu de cela, le nucléoïde se forme par condensation et arrangement fonctionnel à l'aide de protéines architecturales chromosomiques et de molécules d'ARN ainsi que d'un superenroulement d'ADN . La longueur d'un génome varie considérablement (généralement au moins quelques millions de paires de bases) et une cellule peut en contenir plusieurs copies.
Il n'existe pas encore de structure à haute résolution connue d'un nucléoïde bactérien, mais des caractéristiques clés ont été étudiées chez Escherichia coli en tant qu'organisme modèle . Chez E. coli , l'ADN chromosomique est en moyenne négativement superenroulé et plié en boucles plectonémiques , qui sont confinées à différentes régions physiques et diffusent rarement les unes dans les autres. Ces boucles s'organisent spatialement en régions de la taille d'une mégabase appelées macrodomaines, au sein desquelles les sites ADN interagissent fréquemment, mais entre lesquelles les interactions sont rares. L'ADN condensé et organisé spatialement forme un ellipsoïde hélicoïdal qui est confiné radialement dans la cellule. La structure 3D de l'ADN dans le nucléoïde semble varier en fonction des conditions et est liée à l'expression des gènes, de sorte que l'architecture du nucléoïde et la transcription des gènes sont étroitement interdépendantes, s'influençant réciproquement.
Arrière-plan
Chez de nombreuses bactéries, le chromosome est une molécule d'ADN double brin fermée (circulaire) qui code l'information génétique sous une forme haploïde . La taille de l'ADN varie de 500 000 à plusieurs millions de paires de bases (pb) codant de 500 à plusieurs milliers de gènes selon l'organisme. L'ADN chromosomique est présent dans les cellules sous une forme très compacte et organisée appelée nucléoïde (qui signifie semblable à un noyau ), qui n'est pas enfermé dans une membrane nucléaire comme dans les cellules eucaryotes. Le nucléoïde isolé contient 80 % d'ADN, 10 % de protéines et 10 % d'ARN en poids.
La bactérie Gram-négative Escherichia coli est un système modèle pour la recherche sur les nucléoïdes, notamment sur la manière dont l'ADN chromosomique devient le nucléoïde, les facteurs impliqués, ce que l'on sait de sa structure et comment certains aspects structurels de l'ADN influencent l'expression des gènes .
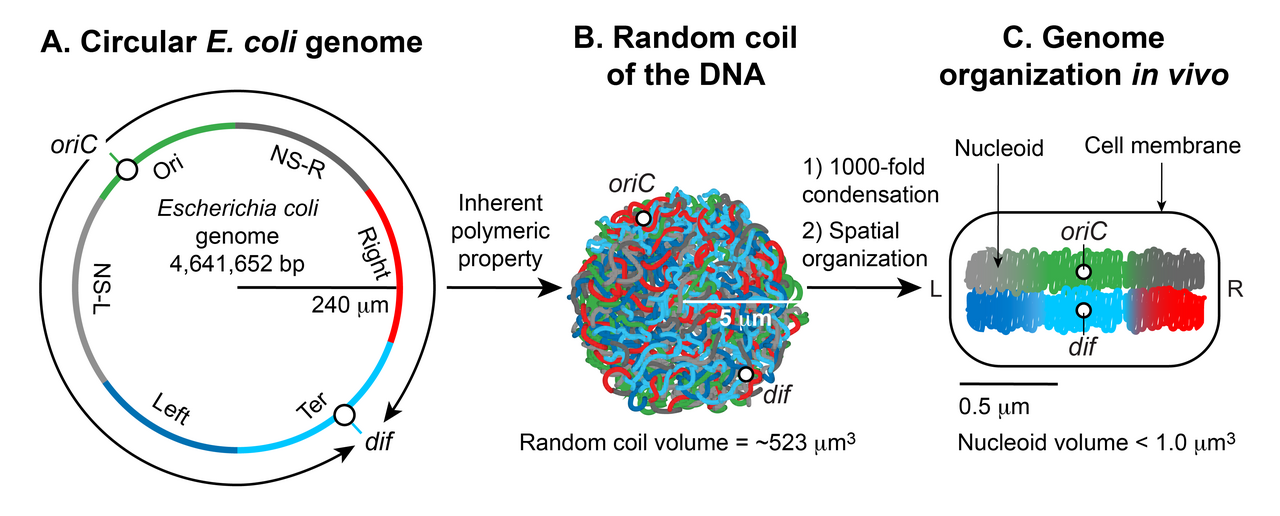
La formation des nucléoïdes comporte deux aspects essentiels : la condensation d'un grand ADN dans un petit espace cellulaire et l'organisation fonctionnelle de l'ADN sous une forme tridimensionnelle. Le chromosome circulaire haploïde d' E. coli est constitué d'environ 4,6 x 10 6 pb. Si l'ADN est relâché sous la forme B , il aurait une circonférence d'environ 1,5 millimètre (0,332 nm x 4,6 x 10 6 ). Cependant, une grande molécule d'ADN telle que l' ADN chromosomique d'E. coli ne reste pas une molécule rigide et droite dans une suspension. Le mouvement brownien génère des courbures et des courbures dans l'ADN. La longueur maximale jusqu'à laquelle un ADN à double hélice reste droit en résistant à la courbure imposée par le mouvement brownien est d'environ 50 nm ou 150 pb, ce qui est appelé la longueur de persistance . Ainsi, l'ADN pur devient sensiblement condensé sans aucun facteur supplémentaire ; à l'équilibre thermique, il prend une forme de bobine aléatoire . La bobine aléatoire de l'ADN chromosomique d'E. coli occuperait un volume (4/3 π r 3 ) d'environ 523 μm 3 , calculé à partir du rayon de giration ( R g = (√N a)/√6) où a est la longueur de Kuhn (2 x longueur de persistance) et N est le nombre de segments de longueur de Kuhn dans l'ADN (longueur totale de l'ADN divisée par a ). Bien que l'ADN soit déjà condensé sous forme de bobine aléatoire, il ne peut toujours pas prendre le volume du nucléoïde qui est inférieur à un micron. Ainsi, la propriété inhérente de l'ADN n'est pas suffisante : des facteurs supplémentaires doivent aider à condenser davantage l'ADN de l'ordre d'environ 10 3 (volume de la bobine aléatoire divisé par le volume du nucléoïde). Le deuxième aspect essentiel de la formation des nucléoïdes est l'agencement fonctionnel de l'ADN. L'ADN chromosomique n'est pas seulement condensé mais également organisé fonctionnellement d'une manière compatible avec les processus de transaction de l'ADN tels que la réplication , la recombinaison , la ségrégation et la transcription . Près de cinq décennies de recherche commençant en 1971, a montré que la forme finale du nucléoïde résulte d'une organisation hiérarchique de l'ADN. À la plus petite échelle (1 kb ou moins), les protéines architecturales de l'ADN associées au nucléoïde condensent et organisent l'ADN en courbant, bouclant, reliant ou en enroulant l'ADN. À une plus grande échelle (10 kb ou plus), l'ADN forme des boucles plectonémiques, une forme tressée de l'ADN induite par un superenroulement. À l'échelle de la mégabase, les boucles plectonémiques fusionnent en six domaines organisés spatialement (macrodomaines), qui sont définis par des interactions physiques plus fréquentes entre les sites d'ADN au sein du même macrodomaine qu'entre différents macrodomaines. Les connexions ADN-ADN à longue et courte portée formées à l'intérieur et entre les macrodomaines contribuent à la condensation et à l'organisation fonctionnelle. Enfin, le nucléoïde est un ellipsoïde hélicoïdal avec des régions d'ADN hautement condensées sur l'axe longitudinal.
Condensation et organisation
Protéines associées aux nucléoïdes (NAP)
Chez les eucaryotes, l'ADN génomique est condensé sous la forme d'un ensemble répétitif de particules d'ADN-protéines appelées nucléosomes .
Un nucléosome est constitué d'environ 146 pb d'ADN enroulé autour d'un complexe octamérique de protéines histones . Bien que les bactéries ne possèdent pas d'histones, elles possèdent un groupe de protéines de liaison à l'ADN appelées protéines associées aux nucléoïdes (NAP) qui sont fonctionnellement analogues aux histones au sens large. Les NAP sont très abondantes et constituent une proportion significative du composant protéique du nucléoïde.
Une caractéristique distinctive des NAP est leur capacité à se lier à l'ADN de manière spécifique (séquence ou structure spécifique) et non spécifique à la séquence. En conséquence, les NAP sont des protéines à double fonction. La liaison spécifique des NAP est principalement impliquée dans la transcription spécifique des gènes , la réplication de l'ADN , la recombinaison et la réparation . Au plus fort de leur abondance, le nombre de molécules de nombreux NAP est plusieurs ordres de grandeur supérieur au nombre de sites de liaison spécifiques dans le génome. Par conséquent, on en déduit que les NAP se lient à l'ADN chromosomique principalement en mode non spécifique à la séquence et que c'est ce mode qui est crucial pour la compaction des chromosomes. La liaison non spécifique à la séquence d'un NAP peut ne pas être complètement aléatoire ; il pourrait y avoir une faible spécificité de séquence et/ou une spécificité structurelle en raison de la conformation de l'ADN dépendante de la séquence ou de la conformation de l'ADN créée par d'autres NAP.
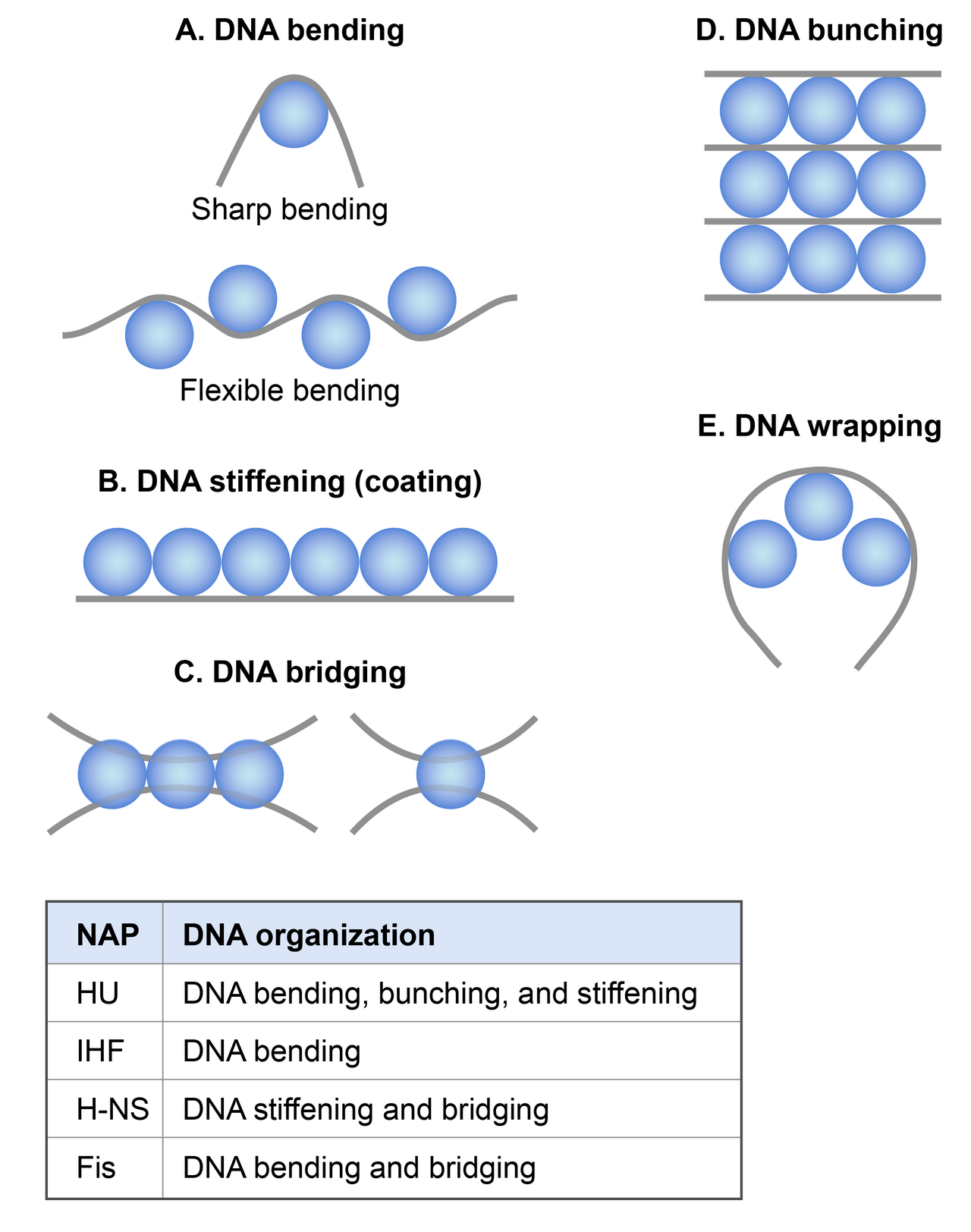
Bien que les mécanismes moléculaires par lesquels les NAP condensent l'ADN in vivo ne soient pas bien compris, d'après les études in vitro approfondies, il semble que les NAP participent au compactage des chromosomes via les mécanismes suivants : les NAP induisent et stabilisent les courbures de l'ADN, contribuant ainsi à la condensation de l'ADN en réduisant la longueur de persistance. Les NAP condensent l'ADN par pontage, enroulement et regroupement qui peuvent se produire entre des segments d'ADN proches ou des segments d'ADN distants du chromosome. Un autre mécanisme par lequel les NAP participent au compactage des chromosomes consiste à contraindre les superenroulements négatifs dans l'ADN, contribuant ainsi à l'organisation topologique du chromosome.
Il existe au moins 12 NAP identifiés dans E. coli, dont les plus étudiés sont HU, IHF, H-NS et Fis. Leur abondance, leurs propriétés de liaison à l'ADN et leur effet sur la condensation et l'organisation de l'ADN sont résumés dans les tableaux ci-dessous.
1 Les données d'abondance (molécules/cellule) ont été tirées de ; Le nombre entre parenthèses est la concentration micromolaire calculée à l'aide de la formule suivante : (nombre d'unités fonctionnelles natives/nombre d'Avogadro) x (1/volume cellulaire en litre) x 10 3 . Le volume cellulaire en litre ( 2 x 10 −15 ) a été déterminé en supposant que le volume de la cellule d'E. coli est de 2 μm 3 .
1 L'affinité de liaison fait référence à la constante de dissociation à l'équilibre (Kd) en unités molaires (M). ND = non déterminé
HU
La protéine de type histone de la souche U93 d'E. coli (HU) est une protéine conservée au cours de l'évolution chez les bactéries. HU existe dans E. coli sous forme d'homo- et d'hétérodimères de deux sous-unités HUα et HUβ partageant 69 % d'identité d'acides aminés. Bien qu'elle soit désignée comme une protéine de type histone, les proches parents fonctionnels de HU chez les eucaryotes sont des protéines du groupe à haute mobilité (HMG) et non des histones. HU est une protéine de liaison à l'ADN non spécifique à la séquence. Elle se lie avec une faible affinité à tout ADN linéaire. Cependant, elle se lie préférentiellement avec une forte affinité à un ADN structurellement déformé. Des exemples de substrats d'ADN déformés comprennent l'ADN cruciforme , l'ADN bombé, l'ADNds contenant une cassure simple brin telle que des entailles , des lacunes ou des fourches . De plus, HU se lie spécifiquement à une boucle d'ADN médiée par des protéines et la stabilise. Dans le mode de liaison à l'ADN structurellement spécifique, HU reconnaît un motif structurel commun défini par des courbures ou des pliures créées par la distorsion, alors qu'il se lie à un ADN linéaire en verrouillant le squelette phosphate. Alors que la liaison structurellement spécifique à haute affinité est nécessaire pour des fonctions spécialisées de HU telles que la recombinaison spécifique au site , la réparation de l'ADN , l'initiation de la réplication de l'ADN et la régulation des gènes, il semble que la liaison générale à faible affinité soit impliquée dans la condensation de l'ADN. Dans l'immunoprécipitation de la chromatine couplée au séquençage de l'ADN ( ChIP-Seq ), HU ne révèle aucun événement de liaison spécifique. Au lieu de cela, il présente une liaison uniforme à travers le génome reflétant vraisemblablement sa liaison principalement faible et non spécifique à la séquence, masquant ainsi la liaison à haute affinité in vivo .
Dans les souches dépourvues de HU, le nucléoïde est « décondensé », ce qui est cohérent avec le rôle de HU dans la compaction de l'ADN. Les études in vitro suivantes suggèrent des mécanismes possibles de la manière dont HU pourrait condenser et organiser l'ADN in vivo . Non seulement HU se lie de manière stable à l'ADN déformé avec des courbures, mais il induit des courbures flexibles même dans un ADN linéaire à une concentration inférieure à 100 nM. En revanche, HU montre l'effet architectural opposé sur l'ADN à des concentrations physiologiquement pertinentes plus élevées. Il forme des filaments de nucléoprotéines rigides provoquant le redressement de l'ADN et non sa courbure. Les filaments peuvent en outre former un réseau d'ADN (regroupement d'ADN) extensible à la fois latéralement et médialement en raison de la multimérisation HU-HU déclenchée par la liaison d'ADN non spécifique à la séquence.
Comment ces comportements de HU sont-ils pertinents à l'intérieur de la cellule ? La formation de filaments nécessite une liaison à haute densité de HU sur l'ADN, un dimère HU par ADN de 9 à 20 pb. Mais il n'y a qu'un dimère HU tous les ~150 pb de l'ADN chromosomique basé sur l'abondance estimée de 30 000 dimères HU par cellule (4600000 pb /30 000). Cela indique que les courbures flexibles sont plus susceptibles de se produire in vivo . La courbure flexible provoquerait une condensation due à une réduction de la longueur de persistance de l'ADN comme le montrent les expériences de pinces magnétiques , qui permettent d'étudier la condensation d'une seule molécule d'ADN par une protéine de liaison à l'ADN. Cependant, en raison de la coopérativité , les filaments et réseaux rigides pourraient se former dans certaines régions du chromosome. La formation de filaments à elle seule n'induit pas de condensation, mais la mise en réseau ou le regroupement de l'ADN peut contribuer de manière substantielle à la condensation en rassemblant des segments chromosomiques distants ou proches.
Fédération internationale de hockey
Le facteur d'intégration de l'hôte (IHF) est structurellement presque identique à l'HU mais se comporte différemment de l'HU sous de nombreux aspects. Contrairement à l'HU, qui se lie préférentiellement à un motif structurel quelle que soit la séquence, l'IHF se lie préférentiellement à une séquence d'ADN spécifique même si la spécificité résulte de la structure et de la déformabilité de l'ADN dépendante de la séquence. La liaison spécifique de l'IHF aux sites apparentés courbe fortement l'ADN de >160 degrés. Une occurrence du motif de séquence apparenté est d'environ 3000 dans le génome d'E. coli . L'abondance estimée de l'IHF dans la phase de croissance est d'environ 6000 dimères par cellule. En supposant qu'un dimère d'IHF se lie à un seul motif et que le nucléoïde contient plus d'un équivalent génomique pendant la phase de croissance exponentielle, la plupart des molécules d'IHF occuperaient des sites spécifiques dans le génome et ne condenseraient probablement l'ADN qu'en induisant une forte courbure.
Outre la liaison préférentielle à une séquence d'ADN spécifique, l'IHF se lie également à l'ADN de manière non spécifique à la séquence avec des affinités similaires à celles de l'HU. Le rôle de la liaison non spécifique de l'IHF dans la condensation de l'ADN semble être essentiel dans la phase stationnaire car l'abondance de l'IHF augmente de cinq fois dans la phase stationnaire et les dimères IHF supplémentaires se lieraient probablement à l'ADN chromosomique de manière non spécifique. Contrairement à l'HU, l'IHF ne forme pas de filaments rigides épais à des concentrations plus élevées. Au lieu de cela, sa liaison non spécifique induit également une courbure de l'ADN, bien que le degré de courbure soit beaucoup plus faible que celui des sites spécifiques et soit similaire à la courbure flexible induite par l'HU dans un ADN linéaire à de faibles concentrations. In vitro , la courbure induite par la liaison non spécifique de l'IHF peut provoquer une condensation de l'ADN et favorise la formation de complexes nucléoprotéiques d'ordre supérieur en fonction des concentrations de chlorure de potassium et de chlorure de magnésium. L’organisation de l’ADN d’ordre supérieur par IHF in vivo n’est pas encore claire.
H-NS
Français Une caractéristique distinctive de la protéine structurante de nucléoïde de type histone ou thermostable (H-NS) des autres NAP est la capacité de passer de la forme homodimérique à des concentrations relativement faibles (< 1 x 10 −5 M) à un état oligomérique à des niveaux plus élevés. En raison de ses propriétés d'oligomérisation, H-NS se propage latéralement le long de l'ADN riche en AT dans une réaction de nucléation , où les sites à haute affinité fonctionnent comme des centres de nucléation. La propagation de H-NS sur l'ADN entraîne deux résultats opposés selon la concentration en magnésium dans la réaction. À une faible concentration en magnésium (< 2 mM), H-NS forme des filaments de nucléoprotéines rigides tandis qu'elle forme des ponts intermoléculaires et intramoléculaires à des concentrations en magnésium plus élevées (> 5 mM). La formation de filaments rigides entraîne un redressement de l'ADN sans condensation tandis que le pontage provoque un repliement substantiel de l'ADN. L'analyse de la liaison H-NS dans le génome par des tests ChIP-Seq a fourni une preuve indirecte de la propagation de H-NS sur l'ADN in vivo . H-NS se lie sélectivement à 458 régions du génome. Bien qu'il ait été démontré que H-NS préfère l'ADN courbé formé par des pistes A répétées dans les séquences d'ADN la base de la liaison sélective est la présence d'un motif de séquence conservé trouvé dans les régions riches en AT. Plus important encore, l'apparition fréquente du motif de séquence dans une région de liaison H-NS qui peut renforcer les interactions coopératives protéine-protéine, et la longueur inhabituellement longue de la région de liaison sont cohérentes avec la propagation de la protéine. Que la formation de filaments ou le pontage de l'ADN soit répandu in vivo dépend de la concentration physiologique de magnésium à l'intérieur de la cellule. Si la concentration en magnésium est uniformément faible (< 5 mM), H-NS formerait des filaments de nucléoprotéines rigides in vivo . Alternativement, s'il y a une distribution inégale de magnésium dans la cellule, cela pourrait favoriser à la fois le pontage et le raidissement de l'ADN, mais dans différentes régions du nucléoïde.
De plus, H-NS est surtout connu comme un silencieux global des gènes qui inhibe préférentiellement la transcription des gènes transférés horizontalement et c'est le filament rigide qui conduit au silençage génique. Pris ensemble, il semble que la formation de filaments rigides soit le résultat le plus probable des interactions H-NS-ADN in vivo qui conduisent au silençage génique mais n'induisent pas de condensation de l'ADN. De manière cohérente, l'absence de H-NS ne modifie pas le volume du nucléoïde. Cependant, il est possible qu'E . coli connaisse une concentration élevée en magnésium dans certaines conditions environnementales. Dans de telles conditions, H-NS peut passer de sa forme induisant des filaments à la forme induisant des ponts qui contribue à la condensation et à l'organisation de l'ADN.
Fis
Le facteur de stimulation d'inversion (Fis) est une protéine de liaison à l'ADN spécifique à une séquence qui se lie à des séquences d'ADN spécifiques contenant un motif symétrique de 15 pb. Comme l'IHF, Fis induit une courbure de l'ADN sur des sites apparentés. La capacité à courber l'ADN est apparente dans la structure de l'homodimère Fis. Un homodimère Fis possède deux motifs hélice-tour-hélice (HTH), un de chaque monomère. Un motif HTH reconnaît généralement le sillon principal de l'ADN. Cependant, la distance entre les hélices de reconnaissance de l'ADN des deux motifs HTH dans l'homodimère Fis est de 25 Å , soit environ 8 Å plus courte que le pas d'un ADN-B canonique , ce qui indique que la protéine doit plier ou tordre l'ADN pour se lier de manière stable. De manière cohérente, la structure cristalline des complexes Fis-ADN montre que la distance entre les hélices de reconnaissance reste inchangée alors que l'ADN se courbe dans la plage de 60 à 75 degrés. Il existe 1464 régions de liaison Fis réparties dans le génome d'E. coli et un motif de liaison, identifié par calcul, correspond au motif connu de 15 pb. La liaison spécifique de Fis à de tels sites induirait des courbures dans l'ADN, contribuant ainsi à la condensation de l'ADN en réduisant la longueur de persistance de l'ADN. De plus, de nombreux sites de liaison Fis se produisent en tandem, comme ceux des promoteurs d'ARN stables, par exemple le promoteur P1 de l'opéron rrnB de l'ARNr . La courbure cohérente de Fis aux sites en tandem est susceptible de créer une micro-boucle d'ADN qui peut contribuer davantage à la condensation de l'ADN.
Outre la liaison spécifique à haute affinité aux sites apparentés, Fis peut se lier à une séquence d'ADN aléatoire. La liaison non spécifique à l'ADN est importante car Fis est aussi abondant que HU dans la phase de croissance . Par conséquent, la plupart des molécules Fis devraient se lier à l'ADN d'une manière non spécifique à la séquence. Des expériences avec des pinces magnétiques montrent que cette liaison non spécifique de Fis peut contribuer à la condensation et à l'organisation de l'ADN. Fis provoque une légère condensation d'une seule molécule d'ADN à < 1 mM, mais induit un repliement substantiel par la formation de boucles d'ADN d'une taille moyenne d'environ 800 pb à > 1 mM. Les boucles dans les expériences avec des pinces magnétiques sont distinctes des micro-boucles créées par la courbure cohérente de l'ADN aux sites apparentés, car elles nécessitent la formation de complexes ADN-protéine à haute densité obtenus par une liaison indépendante de la séquence. Bien que l'apparition de telles boucles in vivo reste à démontrer, une liaison à haute densité de Fis peut se produire in vivo par l'action concertée de liaisons spécifiques et non spécifiques. L'apparition en tandem de sites spécifiques pourrait initier une réaction de nucléation similaire à celle de H-NS, puis une liaison non spécifique conduirait à la formation de réseaux Fis localisés à haute densité. Le pontage entre ces régions localisées peut créer de grandes boucles d'ADN. Fis est exclusivement présent dans la phase de croissance et non dans la phase stationnaire . Ainsi, tout rôle dans la condensation chromosomique par Fis doit être spécifique aux cellules en croissance.
ARN associés aux nucléoïdes (ARNna)
Français Les premières études examinant l'effet du traitement à la RNase A sur des nucléoïdes isolés ont indiqué que l'ARN participait à la stabilisation du nucléoïde à l'état condensé. De plus, le traitement à la RNase A a perturbé les fibres d'ADN en fibres plus fines, comme observé par une microscopie à force atomique du nucléoïde en utilisant la « procédure de lyse sur substrat ». Ces résultats ont démontré la participation de l'ARN dans la structure du nucléoïde, mais l'identité de la ou des molécules d'ARN est restée inconnue jusqu'à récemment. La plupart des études sur l'HU se sont concentrées sur sa liaison à l'ADN. Cependant, l'HU se lie également à l'ARNdb et aux hybrides ARN-ADN avec une affinité plus faible similaire à celle d'un ADNdb linéaire. De plus, l'HU se lie préférentiellement à l'ARN contenant des structures secondaires et à un hybride ARN-ADN dans lequel l'ARN contient une entaille ou un surplomb. Les affinités de liaison de HU avec ces substrats d'ARN sont similaires à celles avec lesquelles il se lie à l'ADN déformé. Une immunoprécipitation d'ARN lié à HU couplée à une étude de transcription inverse et de microarray (RIP-Chip) ainsi qu'une analyse d'ARN à partir de nucléoïdes intacts purifiés ont identifié des molécules d'ARN associées aux nucléoïdes qui interagissent avec HU. Plusieurs d'entre eux sont des ARN non codants, et l'un de ces ARN nommé naRNA4 (nucleoid-associated RNA 4), est codé dans un palindrome extragénique répétitif ( REP325 ). Dans une souche dépourvue de REP325 , le nucléoïde est décondensé comme dans une souche dépourvue de HU. naRNA4 participe très probablement à la condensation de l'ADN en connectant des segments d'ADN en présence de HU. Des études récentes donnent un aperçu du mécanisme moléculaire par lequel naRNA4 établit des connexions ADN-ADN. L'ARN cible les régions d'ADN contenant des structures cruciformes et forme un complexe ARN-ADN essentiel à l'établissement de connexions ADN-ADN. Étonnamment, bien que l'HU contribue à la formation du complexe, il n'est pas présent dans le complexe final, ce qui indique son rôle potentiel de catalyseur (chaperon). La nature du complexe ARN-ADN reste déroutante car la formation du complexe n'implique pas d'appariement de bases Watson/Crick étendu mais est sensible à la RNase H, qui clive l'ARN dans un hybride ARN-ADN et le complexe se lie à un anticorps spécifique aux hybrides ARN-ADN.
Superenroulement
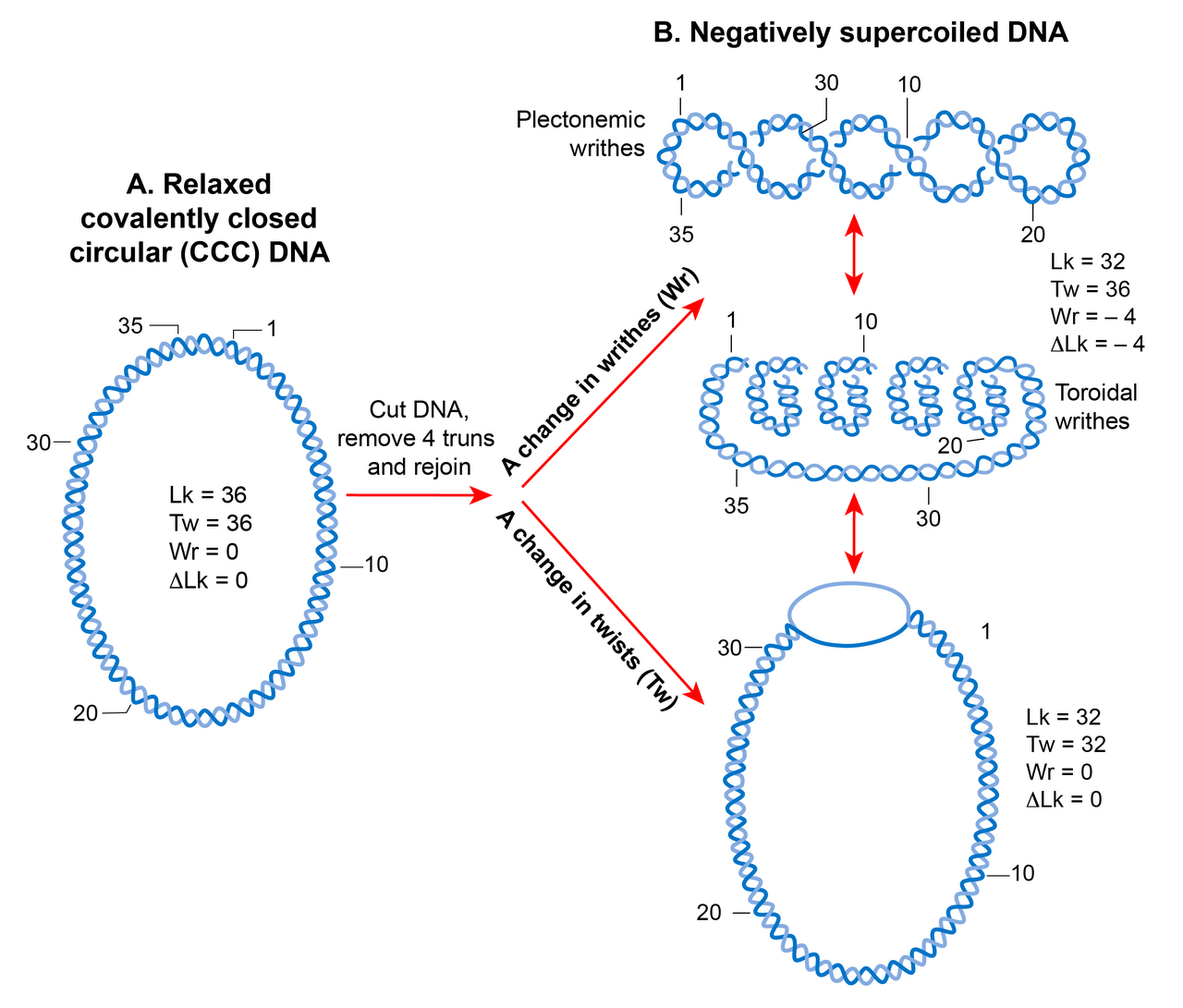
En raison de sa structure hélicoïdale , une molécule d'ADN double brin devient topologiquement contrainte dans la forme circulaire fermée covalente qui élimine la rotation des extrémités libres. Le nombre de fois que les deux brins se croisent dans un ADN topologiquement contraint est appelé le nombre de liaison (Lk), qui est équivalent au nombre de tours ou de torsions hélicoïdaux dans une molécule circulaire. Le Lk d'un ADN topologique reste invariant, quelle que soit la déformation de la molécule d'ADN, tant qu'aucun brin n'est cassé.
Le Lk de l'ADN sous forme relâchée est défini comme Lk 0 . Pour tout ADN, Lk 0 peut être calculé en divisant la longueur (en pb) de l'ADN par le nombre de pb par tour d'hélice. Cela équivaut à 10,4 pb pour l' ADN sous forme B relâchée . Tout écart par rapport à Lk 0 provoque un surenroulement dans l'ADN. Une diminution du nombre de liaisons (Lk<Lk 0 ) crée un surenroulement négatif tandis qu'une augmentation du nombre de liaisons (Lk>Lk 0 ) crée un surenroulement positif.
L'état superenroulé (lorsque Lk n'est pas égal à Lk 0 ) entraîne une transition dans la structure de l'ADN qui peut se manifester par un changement dans le nombre de torsions (négatif < 10,4 pb/tour, positif > 10,4 pb par tour) et/ou par la formation de contorsions , appelées superenroulements. Ainsi, Lk est mathématiquement défini comme une somme dépendante du signe des deux paramètres géométriques, torsion et contorsion. Une mesure quantitative du superenroulement qui est indépendante de la taille des molécules d'ADN est la densité de superenroulement (σ) où σ = ∆Lk/Lk 0 .
Les enroulements peuvent adopter deux structures : plectonème et solénoïde ou toroïde. Une structure plectonémique naît de l'enroulement de l'axe hélicoïdal. Les superenroulements toroïdaux apparaissent lorsque l'ADN forme plusieurs spirales, autour d'un axe et sans se croiser, comme celles d'un cordon téléphonique. Les enroulements sous forme de plectonèmes sont respectivement droitiers et gauchers dans l'ADN positivement ou négativement surenroulé. La sélectivité des superenroulements toroïdaux est opposée à celle des plectonèmes. Les plectonèmes et les superenroulements toroïdaux peuvent être soit sous forme libre, soit retenus sous forme liée par des protéines. Le meilleur exemple de superenroulement toroïdal lié en biologie est le nucléosome eucaryote dans lequel l'ADN s'enroule autour des histones .
Superbobines plectonémiques enEscherichia coli
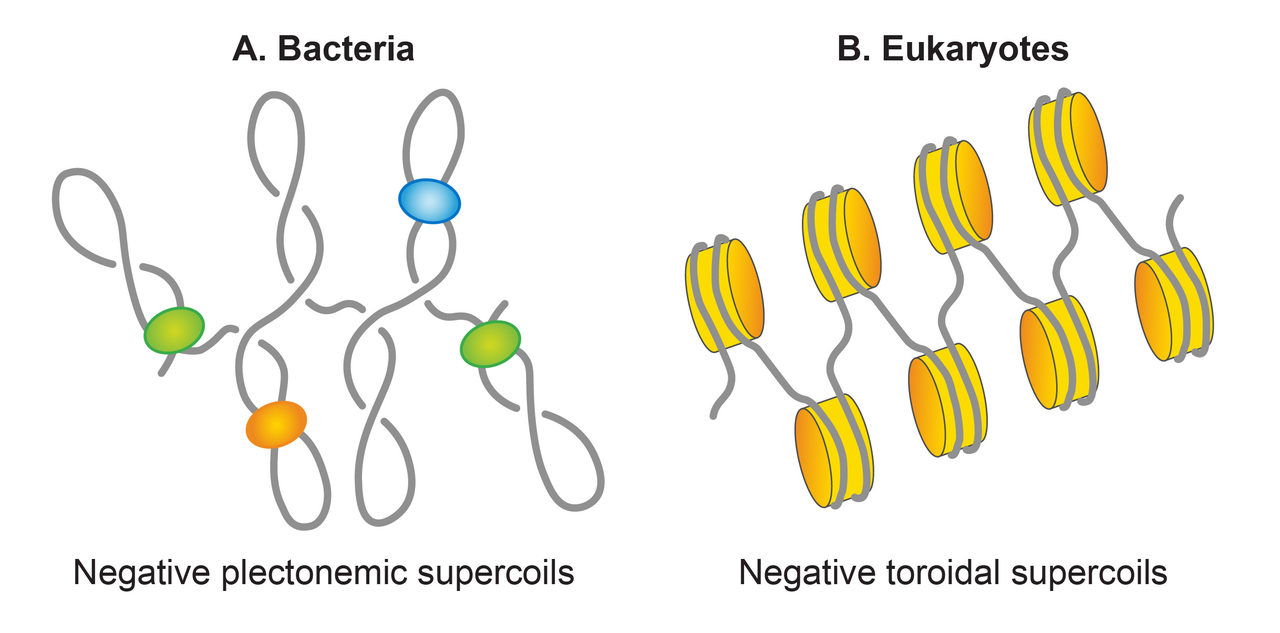
Dans la plupart des bactéries, l'ADN est présent sous forme superenroulée. La nature circulaire du chromosome d'E. coli en fait une molécule topologiquement contrainte qui est principalement superenroulée négativement avec une densité de superenroulement moyenne estimée (σ) de -0,05. chromatine eucaryote , l'ADN se trouve principalement sous la forme toroïdale qui est restreinte et définie par des histones via la formation de nucléosomes. En revanche, dans le nucléoïde d'E. coli , environ la moitié de l'ADN chromosomique est organisée sous la forme de superenroulements plectonémiques libres. L'ADN restant est restreint soit sous la forme plectonémique, soit sous des formes alternatives, y compris, mais sans s'y limiter, la forme toroïdale, par interaction avec des protéines telles que les NAP. Ainsi, les superenroulements plectonémiques représentent un superenroulement efficace du génome d'E. coli qui est responsable de sa condensation et de son organisation. Les superenroulements plectonémiques et toroïdaux facilitent la condensation de l'ADN. La ramification des structures plectonémiques permet une condensation de l'ADN moindre que celle de la structure toroïdale. Une molécule d'ADN de même taille avec des densités de superenroulement égales est plus compacte sous une forme toroïdale que sous une forme plectonémique. En plus de condenser l'ADN, le superenroulement facilite l'organisation de l'ADN. Il favorise le démêlage de l'ADN en réduisant la probabilité de caténation. Le superenroulement permet également de rapprocher deux sites distants de l'ADN, favorisant ainsi une interaction fonctionnelle potentielle entre différents segments d'ADN.
Sources de superenroulement dansEscherichia coli
Trois facteurs contribuent à la génération et au maintien du superenroulement de l'ADN chromosomique chez E. coli : (i) les activités des topoisomérases , (ii) l'acte de transcription et (iii) les NAP.
Topoisomérases
Les topoisomérases sont une catégorie particulière d'enzymes métaboliques de l'ADN qui créent ou suppriment le surenroulement en cassant puis en reliquant les brins d'ADN. E. coli possède quatre topoisomérases. L'ADN gyrase introduit un surenroulement négatif en présence d'ATP et supprime un surenroulement positif en l'absence d'ATP. Dans toutes les formes de vie, l'ADN gyrase est la seule topoisomérase capable de créer un surenroulement négatif et c'est en raison de cette capacité unique que les génomes bactériens possèdent des surenroulements négatifs libres ; l'ADN gyrase est présente dans toutes les bactéries mais absente des eucaryotes supérieurs. En revanche, Topo I s'oppose à l'ADN gyrase en relâchant l'ADN surenroulement négatif. Il existe des preuves génétiques suggérant qu'un équilibre entre les activités opposées de l'ADN gyrase et de Topo I est responsable du maintien d'un niveau stable de superhélicité négative moyenne dans E. coli . Les deux enzymes sont essentielles à la survie d'E. coli . Une souche nulle de topA , le gène codant Topo I, survit uniquement grâce à la présence de mutations suppressives dans les gènes codant l'ADN gyrase. Ces mutations entraînent une activité gyrase réduite, ce qui suggère que l'excès de superenroulement négatif dû à l'absence de Topo I est compensé par une activité de superenroulement négatif réduite de l'ADN gyrase. Topo III est dispensable dans E. coli et n'est pas connu pour avoir un rôle dans le superenroulement chez E. coli. La fonction principale de Topo IV est de résoudre les chromosomes frères. Cependant, il a été démontré qu'il contribue également au niveau d'équilibre du superenroulement négatif en relâchant le superenroulement négatif avec Topo I.
Transcription
Un modèle de domaine de superenroulement jumeau proposé par Liu et Wang a soutenu que le déroulement de la double hélice d'ADN pendant la transcription induit un superenroulement dans l'ADN comme indiqué dans. Selon leur modèle, la transcription de l'ARN polymérase (RNAP) glissant le long de l'ADN force l'ADN à tourner sur son axe hélicoïdal. Un obstacle à la libre rotation de l'ADN pourrait survenir en raison d'une contrainte topologique, provoquant une torsion excessive de l'ADN devant la RNAP (superenroulement positif) et une sous-torsion de l'ADN derrière la RNAP (superenroulement négatif). Il a été découvert qu'une contrainte topologique n'est pas nécessaire car la RNAP génère un couple suffisant pour provoquer un superenroulement même dans un modèle d'ADN linéaire. Si l'ADN est déjà superenroulé négativement, cette action relâche les superenroulements négatifs existants avant de provoquer une accumulation de superenroulements positifs devant la RNAP et introduit davantage de superenroulements négatifs derrière la RNAP. En principe, l'ADN gyrase et Topo I devraient éliminer respectivement les excès de superenroulements positifs et négatifs, mais si le taux d'élongation de l'ARN polymérase dépasse le renouvellement des deux enzymes, la transcription contribue au niveau d'équilibre du superenroulement.
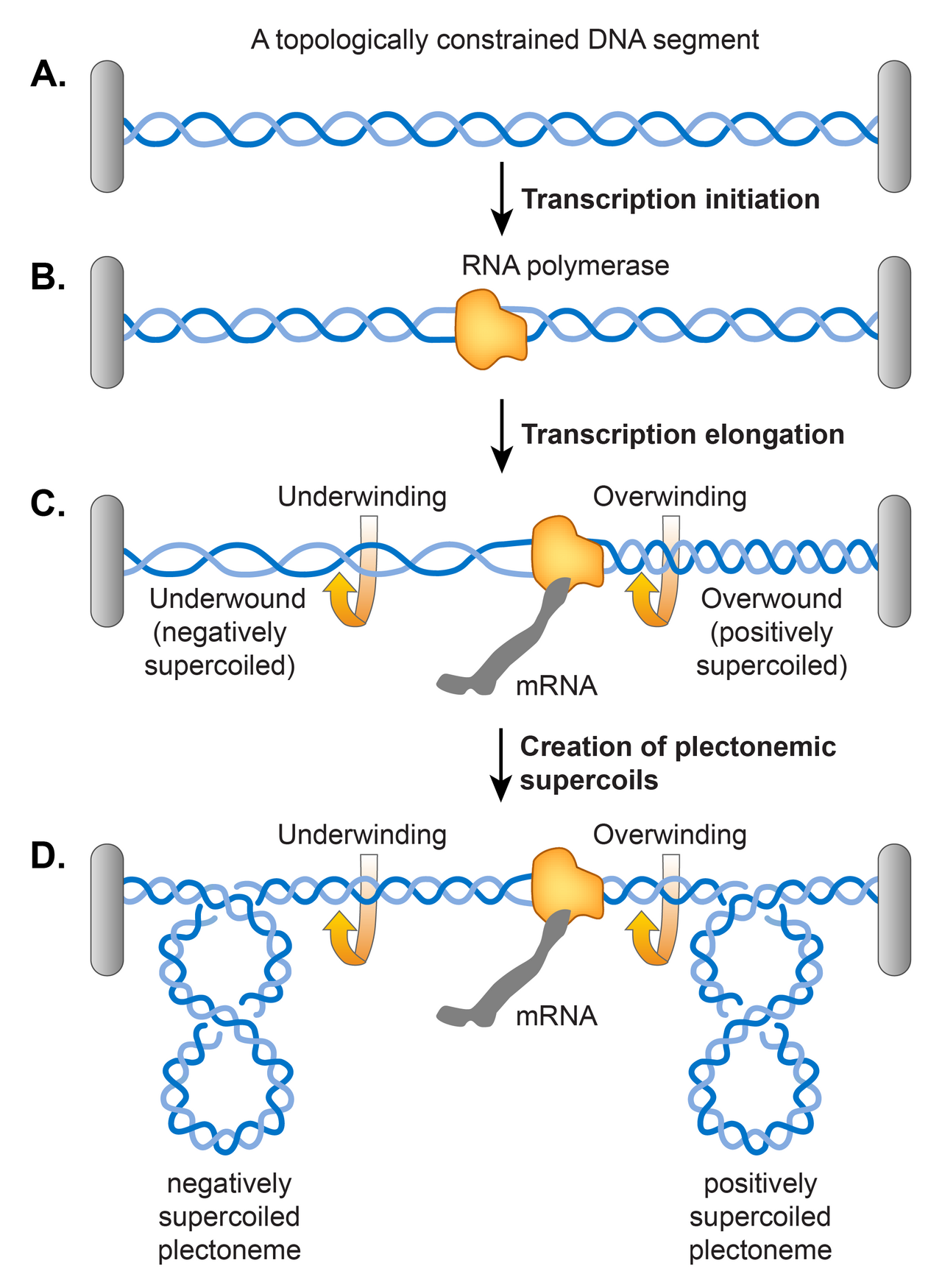
Contrôle du superenroulement par les NAP
Dans la chromatine eucaryote, l'ADN est rarement présent sous forme libre et surenroulée, car les nucléosomes limitent presque tout surenroulement négatif par une liaison étroite de l'ADN aux histones. De même, chez E. coli , les complexes nucléoprotéiques formés par les NAP limitent la moitié de la densité de surenroulement du nucléoïde. En d'autres termes, si une NAP se dissocie d'un complexe nucléoprotéique , l'ADN adopterait la forme libre et plectonémique. Il a été démontré expérimentalement que la liaison à l'ADN de HU, Fis et H-NS limite le surenroulement négatif dans un ADN détendu mais topologiquement contraint. Ils peuvent le faire soit en modifiant le pas hélicoïdal de l'ADN, soit en générant des contorsions toroïdales par courbure et enroulement de l'ADN. Alternativement, les NAP peuvent se lier préférentiellement à d'autres formes d'ADN sous-enroulé et les stabiliser, telles que les structures cruciformes et les plectonèmes ramifiés. Il a été rapporté que Fis organise les plectonèmes ramifiés grâce à sa liaison aux régions de croisement et que HU se lie préférentiellement aux structures cruciformes.
Les NAP régulent également indirectement le surenroulement de l'ADN. Fis peut moduler le surenroulement en réprimant la transcription des gènes codant l'ADN gyrase. Il existe des preuves génétiques suggérant que HU contrôle les niveaux de surenroulement en stimulant l'ADN gyrase et en réduisant l'activité de Topo I. À l'appui des études génétiques, il a été démontré que HU stimule la décaténation de l'ADN catalysée par l'ADN gyrase in vitro . On ne sait pas clairement comment mécaniquement HU module les activités de la gyrase et de Topo I. HU pourrait interagir physiquement avec l'ADN gyrase et Topo I ou les activités d'organisation de l'ADN de HU telles que la courbure de l'ADN peuvent faciliter ou inhiber l'action de l'ADN gyrase et de Topo I respectivement.
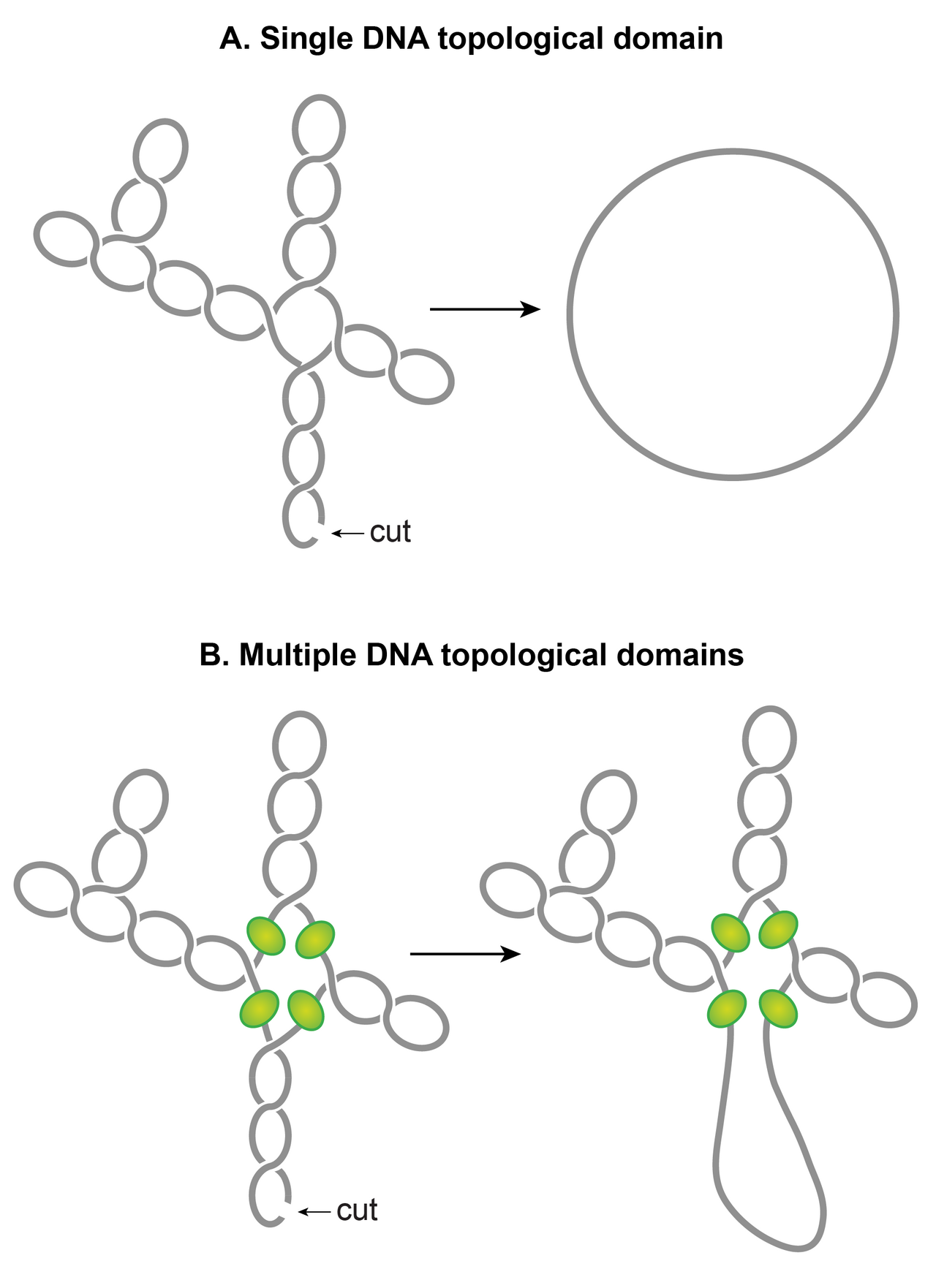
Les superbobines plectonémiques s'organisent en plusieurs domaines topologiques
L'une des caractéristiques frappantes du nucléoïde est que les superenroulements plectonémiques sont organisés en plusieurs domaines topologiques. En d'autres termes, une seule coupure dans un domaine ne détendra que ce domaine et pas les autres. Un domaine topologique se forme en raison d'une barrière de superenroulement-diffusion. Des études indépendantes employant différentes méthodes ont rapporté que les domaines topologiques ont une taille variable allant de 10 à 400 kb. Un placement aléatoire des barrières couramment observé dans ces études semble expliquer la grande variabilité de la taille des domaines.
Bien que les identités des barrières de domaine restent à établir, les mécanismes possibles responsables de la formation des barrières incluent : (i) Une barrière de domaine pourrait se former lorsqu'une protéine ayant la capacité de restreindre les superenroulements se lie simultanément à deux sites distincts sur le chromosome, formant ainsi une boucle ou un domaine d'ADN topologiquement isolé. Il a été démontré expérimentalement que la boucle médiée par les protéines dans l'ADN superenroulé peut créer un domaine topologique. Les NAP tels que H-NS et Fis sont des candidats potentiels, en fonction de leurs capacités de boucle d'ADN et de la distribution de leurs sites de liaison. (ii) Les éléments de mosaïque intercalés bactériens (BIME) apparaissent également comme des candidats potentiels pour les barrières de domaine. Les BIME sont des séquences répétées palindromiques que l'on trouve généralement entre les gènes. Il a été démontré qu'un BIME empêche la diffusion du superenroulement dans une cassette topologique conçue synthétiquement insérée dans le chromosome d'E. coli . Il existe environ 600 BIME répartis dans le génome, divisant peut-être le chromosome en 600 domaines topologiques. (iii) Les barrières pourraient également résulter de la fixation de l'ADN à la membrane cellulaire par l'intermédiaire d'une protéine qui se lie à la fois à l'ADN et à la membrane ou par la transcription naissante et la traduction de protéines ancrées à la membrane. (iv) L'activité de transcription peut générer des barrières de superenroulement-diffusion. Il a été démontré qu'une RNAP à transcription active bloque la dissipation des superenroulements plectonémiques, formant ainsi une barrière de superenroulement-diffusion.
Dynamique des nucléoïdes en fonction de la phase de croissance
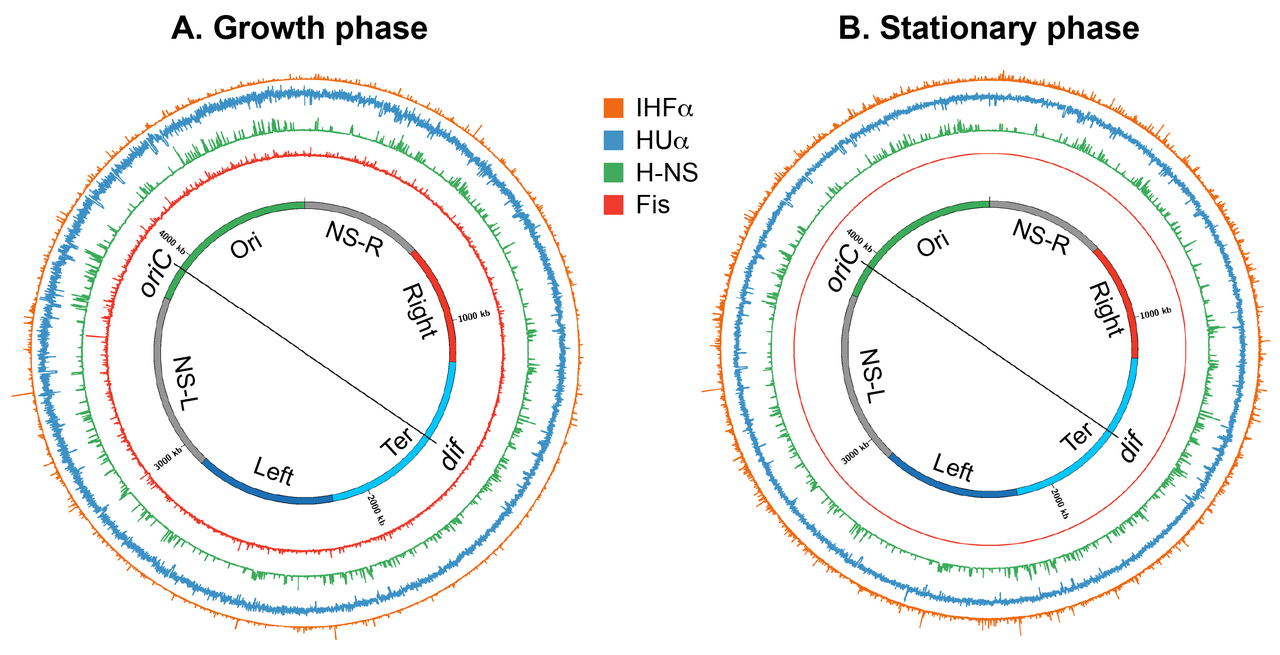
Le nucléoïde se réorganise dans les cellules en phase stationnaire, ce qui suggère que la structure du nucléoïde est hautement dynamique, déterminée par l'état physiologique des cellules. Une comparaison des cartes de contact à haute résolution du nucléoïde a révélé que les contacts à longue portée dans le macrodomaine Ter ont augmenté dans la phase stationnaire , par rapport à la phase de croissance . De plus, les limites CID dans la phase stationnaire étaient différentes de celles trouvées dans la phase de croissance. Enfin, la morphologie du nucléoïde subit une transformation massive pendant la phase stationnaire prolongée ; le nucléoïde présente des structures toroïdales ordonnées.
Les changements spécifiques de la phase de croissance dans la structure du nucléoïde pourraient être provoqués par un changement des niveaux de protéines architecturales de l'ADN associées au nucléoïde (les NAP et les sous-unités Muk), le superenroulement et l'activité de transcription. L'abondance des NAP et des sous-unités Muk change en fonction du cycle de croissance bactérienne. Fis et la protéine de liaison à l'ADN induite par la famine Dps, une autre NAP, sont presque exclusivement présentes respectivement dans la phase de croissance et la phase stationnaire. Les niveaux de Fis augmentent lors de l'entrée dans la phase exponentielle, puis diminuent rapidement alors que les cellules sont encore dans la phase exponentielle, atteignant des niveaux indétectables dans la phase stationnaire. Alors que les niveaux de Fis commencent à diminuer, les niveaux de Dps commencent à augmenter et atteignent un maximum dans la phase stationnaire. Une transition spectaculaire dans la structure du nucléoïde observée dans la phase stationnaire prolongée a été principalement attribuée à Dps. Il forme des assemblages ADN/ cristallins qui agissent pour protéger le nucléoïde des agents endommageant l'ADN présents pendant la famine.
HU, IHF et H-NS sont présents à la fois en phase de croissance et en phase stationnaire. Cependant, leur abondance change de manière significative de telle sorte que HU et Fis sont les NAP les plus abondants en phase de croissance, tandis que IHF et Dps deviennent les NAP les plus abondants en phase stationnaire. HUαα est la forme prédominante en phase exponentielle précoce, tandis que la forme hétérodimérique prédomine en phase stationnaire, avec des quantités mineures d'homodimères. Cette transition a des conséquences fonctionnelles sur la structure des nucléoïdes, car les deux formes semblent organiser et condenser l'ADN différemment ; les homo- et les hétérodimères forment des filaments, mais seul l'homodimère peut rassembler plusieurs segments d'ADN pour former un réseau d'ADN. Le nombre de copies de MukB augmente de moitié en phase stationnaire. Une augmentation du nombre de molécules MukB pourrait avoir une influence sur la processivité du complexe MukBEF en tant que facteur d'extrusion de boucle d'ADN, ce qui entraînerait un nombre plus grand ou plus important de boucles.
Le superenroulement peut agir de manière concertée avec les protéines architecturales de l'ADN pour réorganiser le nucléoïde. Le niveau global de superenroulement diminue dans la phase stationnaire et le superenroulement présente un schéma différent au niveau régional. Les changements dans le superenroulement peuvent modifier l'organisation topologique du nucléoïde. De plus, comme une région chromosomique à forte activité de transcription forme une limite CID, les changements dans l'activité de transcription au cours des différentes phases de croissance pourraient modifier la formation des limites CID et donc l'organisation spatiale du nucléoïde. Il est possible que les changements dans les limites CID observés dans la phase stationnaire soient dus à la forte expression d'un ensemble différent de gènes dans la phase stationnaire par rapport à la phase de croissance.
Structure nucléoïde et expression génétique
NAP et expression génétique
La structure du chromosome d'E. coli et l'expression des gènes semblent s'influencer mutuellement. D'une part, une corrélation entre une limite CID et une activité de transcription élevée indique que l'organisation des chromosomes est déterminée par la transcription. D'autre part, la structure 3D de l'ADN au sein du nucléoïde à toutes les échelles peut être liée à l'expression des gènes. Tout d'abord, il a été démontré que la réorganisation de l'architecture 3D du nucléoïde chez E. coli peut moduler de manière dynamique le modèle de transcription cellulaire. Un mutant de HUa a rendu le nucléoïde très condensé par une superhélicité positive accrue de l'ADN chromosomique. Par conséquent, de nombreux gènes ont été réprimés et de nombreux gènes quiescents ont été exprimés. En outre, il existe de nombreux cas spécifiques dans lesquels des changements architecturaux locaux médiés par des protéines altèrent la transcription des gènes. Par exemple, la formation de filaments nucléoprotéiques rigides par H-NS bloque l'accès de l'ARN polymérase au promoteur, empêchant ainsi la transcription des gènes. Par le biais du silençage génique, H-NS agit comme un répresseur global inhibant préférentiellement la transcription des gènes transférés horizontalement. Dans un autre exemple, la liaison spécifique de HU à l' opéron gal facilite la formation d'une boucle d'ADN qui maintient l' opéron gal réprimé en l'absence de l'inducteur. La micro-boucle d'ADN topologiquement distincte créée par la courbure cohérente de l'ADN par Fis au niveau des promoteurs d'ARN stables active la transcription. La courbure de l'ADN par IHF contrôle différemment la transcription à partir des deux promoteurs en tandem de l' opéron ilvGMEDA dans E. coli . Des changements topologiques spécifiques par les NAP régulent non seulement la transcription des gènes, mais sont également impliqués dans d'autres processus tels que l'initiation de la réplication de l'ADN, la recombinaison et la transposition. Contrairement à la régulation spécifique des gènes, il reste à déterminer comment la structure chromosomique d'ordre supérieur et sa dynamique influencent l'expression des gènes à l'échelle moléculaire.
Superenroulement de l'ADN et expression génétique
Il existe une interconnexion bidirectionnelle entre le superenroulement de l'ADN et la transcription des gènes. Le superenroulement négatif de la région promotrice peut stimuler la transcription en facilitant la fusion du promoteur et en augmentant l'affinité de liaison à l'ADN d'un régulateur protéique. Les explosions stochastiques de transcription semblent être une caractéristique générale des gènes hautement exprimés, et les niveaux de superenroulement du modèle d'ADN contribuent à l'explosion transcriptionnelle. Selon le modèle de domaine de superenroulement jumeau, la transcription d'un gène peut influencer la transcription d'autres gènes proches par l'intermédiaire d'un relais de superenroulement. Un tel exemple est l'activation du promoteur leu-500 . Le superenroulement n'intervient pas seulement dans les changements spécifiques aux gènes, mais également dans les changements à grande échelle de l'expression des gènes. L'organisation topologique du nucléoïde pourrait permettre l'expression indépendante de gènes sensibles au superenroulement dans différents domaines topologiques. Une carte à l'échelle du génome du superenroulement non restreint a montré que les régions génomiques ont des densités de superenroulement à l'état stable différentes, indiquant que le niveau de superenroulement diffère dans les domaines topologiques individuels. Par conséquent, un changement dans le superenroulement peut entraîner une expression génétique spécifique au domaine, en fonction du niveau de superenroulement dans chaque domaine.
L'effet du superenroulement sur l'expression des gènes peut être médié par des NAP qui influencent directement ou indirectement le superenroulement. L'effet de HU sur l'expression des gènes semble impliquer un changement dans le superenroulement et peut-être une organisation d'ADN d'ordre supérieur. Une corrélation positive entre la liaison de l'ADN gyrase et la régulation positive des gènes causée par l'absence de HU suggère que les changements dans le superenroulement sont responsables de l'expression différentielle. HU s'est également avéré responsable d'un effet positionnel sur l'expression des gènes en isolant les unités transcriptionnelles en contraignant le superenroulement induit par la transcription. Des mutations ponctuelles dans HUa ont radicalement modifié le profil d'expression des gènes d' E. coli, altérant sa morphologie , sa physiologie et son métabolisme . En conséquence, la souche mutante était plus invasive dans les cellules de mammifères. Cet effet spectaculaire était concomitant avec la compaction des nucléoïdes et une augmentation du superenroulement positif. La protéine mutante était un octamère, contrairement au dimère de type sauvage. Il enroule l'ADN sur sa surface de manière droite, limitant les superenroulements positifs contrairement au HU de type sauvage. Ces études montrent que les substitutions d'acides aminés dans le HU peuvent avoir un effet spectaculaire sur la structure du nucléoïde, ce qui entraîne à son tour des changements phénotypiques importants.
Étant donné que MukB et HU sont devenus des acteurs essentiels dans les interactions à longue distance de l'ADN, il sera intéressant de comparer l'effet de chacune de ces deux protéines sur l'expression globale des gènes. Bien que HU semble contrôler l'expression des gènes en modulant la densité de superenroulement, le mécanisme moléculaire exact reste inconnu et l'impact de MukB sur l'expression des gènes n'a pas encore été analysé.
Organisation spatiale
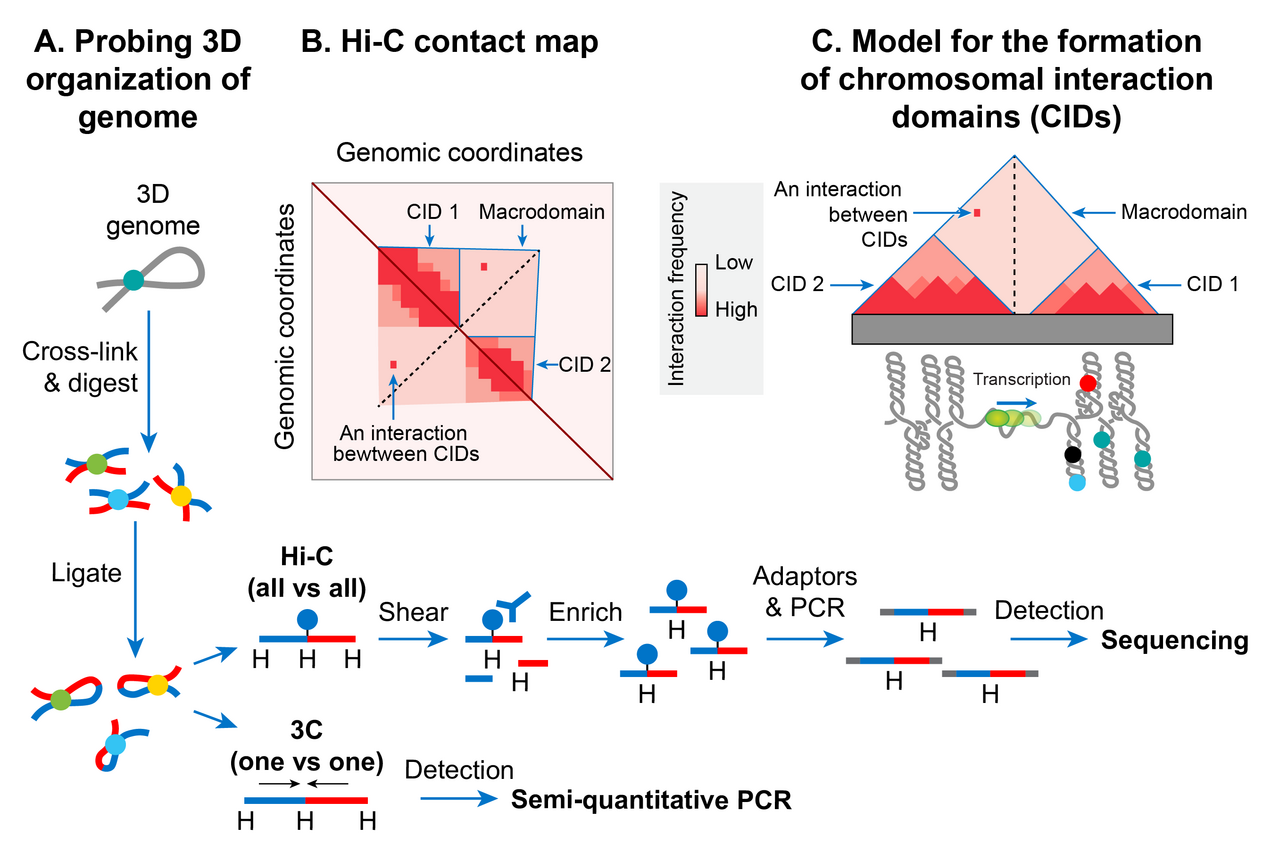
Domaines d'interaction chromosomique
Ces dernières années, l'avènement d'une méthode moléculaire appelée capture de conformation chromosomique (3C) a permis d'étudier une organisation spatiale à haute résolution des chromosomes chez les bactéries et les eucaryotes. La 3C et sa version couplée au séquençage profond (Hi-C) déterminent la proximité physique, le cas échéant, entre deux loci génomiques dans l'espace 3D. Une carte de contact à haute résolution des chromosomes bactériens, y compris le chromosome d'E. coli, a révélé qu'un chromosome bactérien est segmenté en de nombreuses régions hautement interagissantes appelées domaines d'interaction chromosomique (CID). Les CID sont équivalents aux domaines d'association topologique (TAD) observés dans de nombreux chromosomes eucaryotes, suggérant que la formation de CID est un phénomène général d'organisation du génome. Deux caractéristiques définissent les CID ou les TAD. Premièrement, les régions génomiques d'un CID interagissent physiquement entre elles plus fréquemment qu'avec les régions génomiques extérieures à ce CID ou avec celles d'un CID voisin. Deuxièmement, la présence d'une frontière entre les CID empêche les interactions physiques entre les régions génomiques de deux CID voisins.
Le chromosome d'E. coli se compose de 31 CID en phase de croissance. La taille des CID varie de 40 à ~300 kb. Il semble qu'une barrière de superenroulement-diffusion responsable de la ségrégation des boucles d'ADN plectonémiques en domaines topologiques fonctionne comme une limite de CID chez E. coli et de nombreuses autres bactéries. En d'autres termes, la présence d'une barrière de superenroulement-diffusion définit la formation de CID. Les résultats du sondage Hi-C des chromosomes chez E. coli , Caulobacter crescentus et Bacillus subtilis convergent vers un modèle selon lequel les CID se forment parce que la boucle plectonémique associée aux activités d'organisation de l'ADN des NAP favorise les interactions physiques entre les loci génomiques, et une limite de CID consiste en une région sans plectonème (PFR) qui empêche ces interactions. Un PFR est créé en raison d'une activité de transcription élevée car le déroulement hélicoïdal de l'ADN par la transcription active de l'ARN polymérase restreint les superenroulements plectonémiques. En conséquence, la dissipation des superenroulements est également bloquée, créant une barrière de diffusion par superenroulement. Une preuve indirecte de ce modèle provient d'une observation selon laquelle les CID des chromosomes bactériens, y compris le chromosome E. coli, présentent des gènes hautement transcrits à leurs limites, indiquant un rôle de la transcription dans la formation d'une limite CID. Une preuve plus directe provient d'une découverte selon laquelle le placement d'un gène hautement transcrit à une position où aucune limite n'était présente créait une nouvelle limite CID dans le chromosome C. crescentus . Cependant, toutes les limites CID ne sont pas corrélées avec des gènes hautement transcrits dans le chromosome E. coli , ce qui suggère que d'autres facteurs inconnus sont également responsables de la formation de limites CID et de barrières de diffusion par superenroulement.
Macrodomaines
Les boucles d'ADN plectonémiques organisées en domaines topologiques ou CID semblent se regrouper davantage pour former de grands domaines spatialement distincts appelés macrodomaines (MD). Chez E. coli, les MD ont été initialement identifiés comme de grands segments du génome dont les marqueurs d'ADN se localisaient ensemble (colocalisés) dans des études d'hybridation in situ en fluorescence (FISH). Une grande région génomique (~1-Mb) couvrant le locus oriC (origine de la réplication du chromosome) était colocalisée et a été appelée macrodomaine Ori. De même, une grande région génomique (~1-Mb) couvrant la région du terminus de réplication ( ter ) était colocalisée et a été appelée macrodomaine Ter. Les MD ont ensuite été identifiés en fonction de la fréquence à laquelle des paires de sites lambda att insérés à divers endroits distants du chromosome se recombinaient entre eux. Dans cette méthode basée sur la recombinaison, un MD a été défini comme une grande région génomique dont les sites d'ADN peuvent principalement se recombiner entre eux, mais pas avec ceux situés en dehors de ce MD. La méthode basée sur la recombinaison a confirmé les MD Ori et Ter qui ont été identifiés dans les études FISH et a identifié deux MD supplémentaires.
Les deux MD supplémentaires ont été formés par les régions supplémentaires d'environ 1 Mb flanquant le Ter et ont été appelés Gauche et Droite. Ces quatre MD (Ori, Ter, Gauche et Droite) composaient la majeure partie du génome, à l'exception de deux régions génomiques flanquant l'Ori. Ces deux régions (NS-L et NS-R) étaient plus flexibles et non structurées par rapport à un MD, car les sites d'ADN qu'elles contenaient se recombinaient avec les sites d'ADN situés dans les MD des deux côtés. La position génétique de l'oriC semble dicter la formation des MD, car le repositionnement de l' oriC par manipulation génétique entraîne la réorganisation des MD. Par exemple, les régions génomiques les plus proches de l' oriC se comportent toujours comme un NS quelle que soit la séquence d'ADN et les régions plus éloignées se comportent toujours comme des MD.
La technique Hi-C a en outre confirmé une organisation spatiale hiérarchique des CID sous la forme de macrodomaines. En d'autres termes, les CID d'un macrodomaine interagissaient physiquement les uns avec les autres plus fréquemment qu'avec les CID d'un macrodomaine voisin ou avec des loci génomiques en dehors de ce macrodomaine. Les données Hi-C ont montré que le chromosome d'E. coli se divisait en deux domaines distincts. La région entourant ter formait un domaine isolé qui chevauchait le MD Ter précédemment identifié. Les contacts ADN-ADN dans ce domaine ne se produisaient que dans la plage allant jusqu'à ~280 kb. Le reste du chromosome formait un seul domaine dont les loci génomiques présentaient des contacts dans la plage de >280 kb. Alors que la plupart des contacts dans ce domaine étaient limités à une distance maximale d'environ 500 kb, il y avait deux régions lâches dont les loci génomiques formaient des contacts à des distances encore plus grandes (jusqu'à environ 1 Mb). Ces régions lâches correspondaient aux régions flexibles et moins structurées précédemment identifiées (NS). Les limites du domaine isolé englobant ter et les deux régions lâches identifiées par la méthode Hi-C ont segmenté l'ensemble du chromosome en six régions qui correspondent aux quatre MD et aux deux régions NS définies par des tests basés sur la recombinaison.
Protéines qui stimulent la formation des macrodomaines
MatP
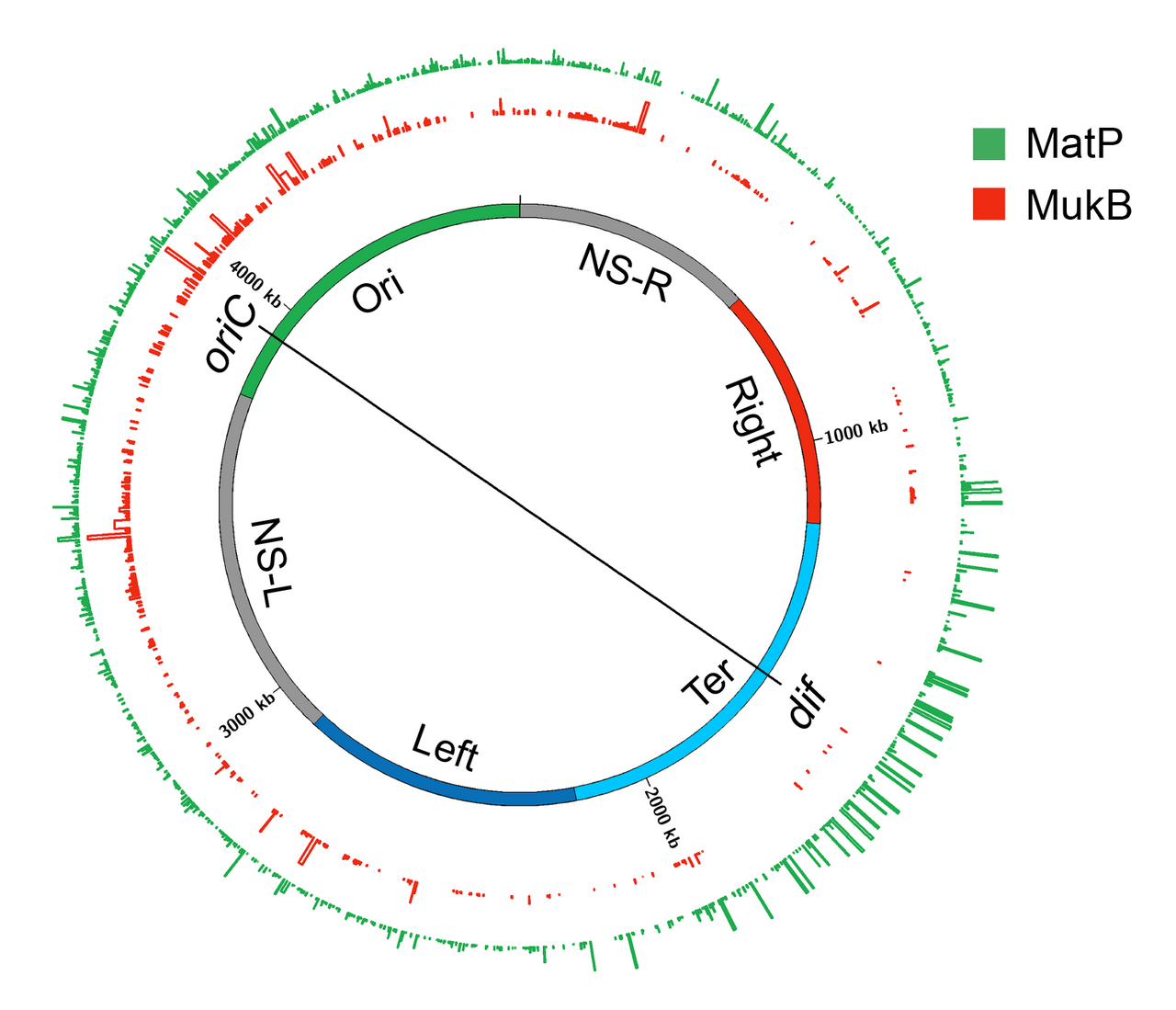
La recherche de protéines responsables de la formation des macrodomaines a conduit à l'identification de la protéine Macrodomain Ter (MatP). MatP se lie presque exclusivement dans le domaine Ter MD en reconnaissant un motif de 13 pb appelé séquence macrodomaine ter ( matS ). Il y a 23 sites matS présents dans le domaine Ter, en moyenne il y a un site tous les 35 kb. D'autres preuves de la liaison de MatP dans le domaine Ter proviennent de l'imagerie par fluorescence de MatP. Des foyers discrets de MatP ont été observés qui étaient co-localisés avec des marqueurs d'ADN du domaine Ter. Un fort enrichissement du signal ChIP-Seq dans le domaine Ter MD corrobore également la liaison préférentielle de MatP à ce domaine.
MatP condense l'ADN dans le domaine Ter car l'absence de MatP a augmenté la distance entre deux marqueurs d'ADN fluorescents situés à 100 kb de distance dans le domaine Ter. De plus, MatP est un acteur essentiel dans l'isolation du domaine Ter du reste du chromosome. Il favorise les contacts ADN-ADN dans le domaine Ter mais empêche les contacts entre les loci d'ADN du domaine Ter et ceux des régions flanquantes. Comment MatP condense-t-il l'ADN et favorise-t-il les contacts ADN-ADN ? Les résultats expérimentaux sont contradictoires. MatP peut former une boucle d'ADN entre deux sites matS in vitro et son activité de boucle d'ADN dépend de la tétramérisation de MatP. La tétramérisation se produit via des interactions en spirale entre deux molécules de MatP liées à l'ADN. Un modèle évident basé sur des résultats in vitro est que MatP favorise les contacts ADN-ADN in vivo en reliant les sites matS . Cependant, bien que MatP ait connecté des sites distants dans les études Hi-C, il n'a pas connecté spécifiquement les sites matS . De plus, un mutant MatP incapable de former des tétramères se comportait comme le type sauvage. Ces résultats vont à l'encontre du modèle de pontage matS pour l'organisation Ter, laissant le mécanisme d'action de MatP insaisissable. Une possibilité est que MatP se propage aux segments d'ADN proches à partir de son site de liaison matS primaire et relie des sites distants via un mécanisme qui ne dépend pas de la tétramérisation.
MukBEF
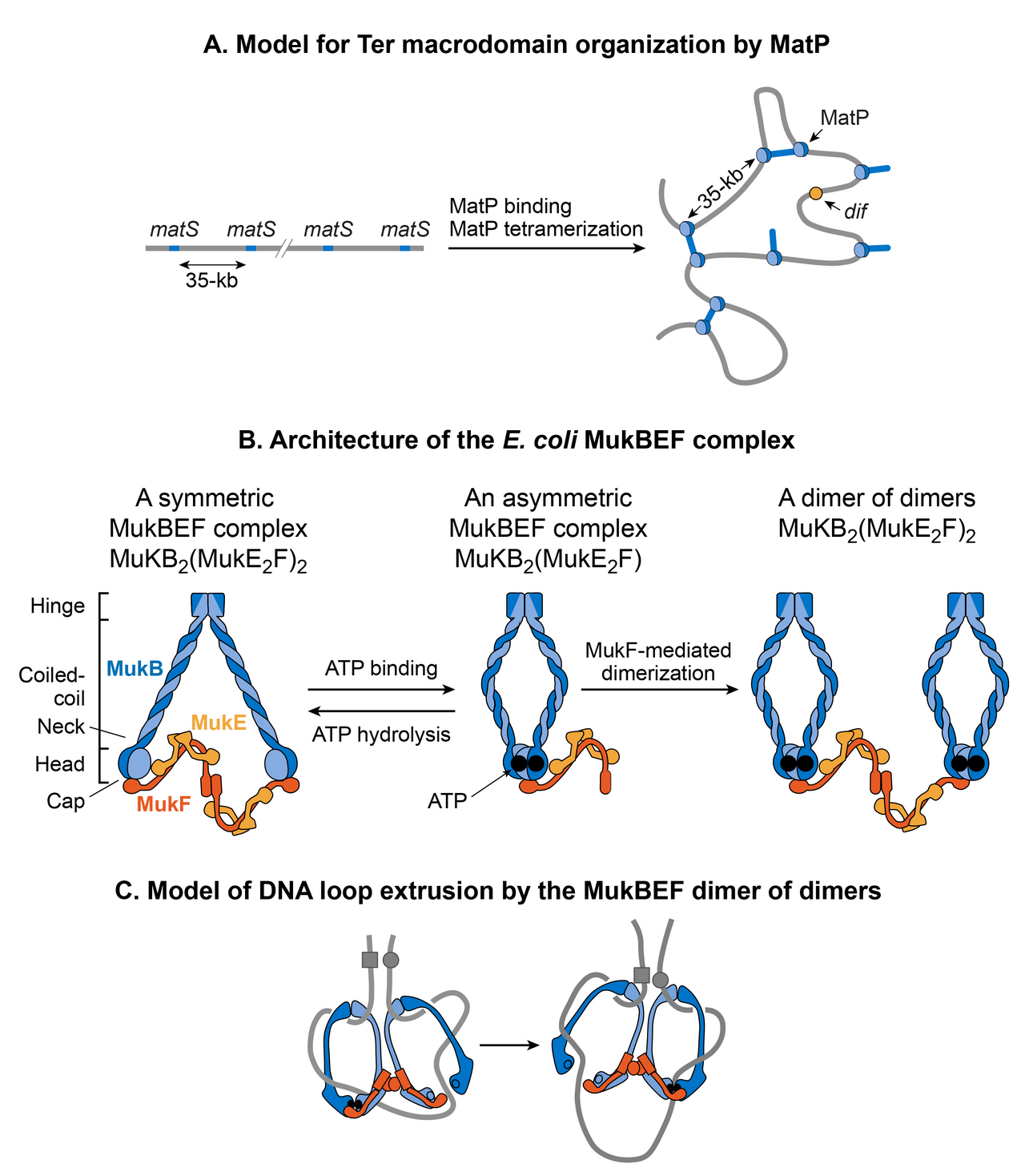
MukB appartient à une famille d'ATPases appelées protéines de maintien structurel des chromosomes (SMC), qui participent à l'organisation des chromosomes d'ordre supérieur chez les eucaryotes. Deux monomères de MukB s'associent via une interaction continue en spirale antiparallèle formant une tige rigide de 100 nm de long. Une région charnière flexible se produit au milieu de la tige. En raison de la flexibilité de la région charnière, MukB adopte une forme en V caractéristique de la famille SMC. Les sous-unités non SMC associées à MukB sont MukE et MukF. L'association ferme la formation en V, ce qui donne lieu à de grandes structures en forme d'anneau. MukE et MukF sont codés avec MukB dans le même opéron chez E. coli . La suppression de l'une ou l'autre des sous-unités entraîne le même phénotype, ce qui suggère que le complexe MukBEF est l'unité fonctionnelle in vivo . Les activités de liaison à l’ADN du complexe résident dans la sous-unité MukB, tandis que MukE et MukF modulent l’activité MukB.
Le complexe MukBEF, avec Topo IV, est nécessaire à la décaténation et au repositionnement des oriC nouvellement répliqués . Le rôle de MukBEF n'est pas limité pendant la réplication de l'ADN. Il organise et condense l'ADN même dans les cellules non réplicatives. La récente carte de conformation chromosomique à haute résolution de la souche E. coli appauvrie en MukB révèle que MukB participe à la formation d'interactions ADN-ADN sur l'ensemble du chromosome, sauf dans le domaine Ter. Comment MukB est-il empêché d'agir dans le domaine Ter ? MatP interagit physiquement avec MukB, empêchant ainsi MukB de se localiser dans le domaine Ter. Ceci est évident dans la liaison à l'ADN de MatP et MukB dans le domaine Ter. La liaison à l'ADN de MatP est enrichie dans le domaine Ter, tandis que la liaison à l'ADN de MukB est réduite par rapport au reste du génome. De plus, dans une souche déjà dépourvue de MatP, l'absence de MukB provoque une réduction des contacts ADN dans tout le chromosome, y compris le domaine Ter. Ce résultat concorde avec l'idée selon laquelle MatP déplace MukB du domaine Ter.
Comment fonctionne le complexe MukBEF pour organiser le chromosome d'E. coli ? Selon l'opinion actuelle, les complexes SMC organisent les chromosomes en extrudant des boucles d'ADN. Les complexes SMC se déplacent le long de l'ADN pour extruder des boucles de manière cis (sur la même molécule d'ADN), la taille des boucles dépendant de la processivité du complexe. Les complexes SMC de différents organismes diffèrent dans le mécanisme d'extrusion de boucle. La microscopie à fluorescence à molécule unique de MukBEF dans E. coli suggère que l'unité fonctionnelle minimale in vivo est un dimère de dimères. Cette unité est formée par la jonction de deux complexes MukBEF liés à l'ATP par le biais d'une dimérisation médiée par MukF. MukBEF se localise dans la cellule sous forme de 1 à 3 clusters qui sont allongés parallèlement à l'axe long de la cellule. Chaque cluster contient en moyenne ~ 8 à 10 dimères de dimères. Selon le modèle actuel, le MukBEF extrude les boucles d'ADN de manière « escaladant un rocher ». Un dimère de dimères libère un segment d'ADN et capture un nouveau segment d'ADN sans se dissocier du chromosome. Outre la boucle d'ADN, un lien entre le superenroulement négatif et la fonction MukBEF in vivo ainsi que la capacité de la sous-unité MukB à contraindre les superenroulements négatifs in vitro suggèrent que le MukBEF organise l'ADN en générant des superenroulements.
Rôle des NAP et des naRNA
En plus de contribuer à la compaction des chromosomes en courbant, en pontant et en bouclant l'ADN à une plus petite échelle (~1 kb), les NAP participent à la condensation et à l'organisation de l'ADN en favorisant les contacts ADN-ADN à longue portée. Deux NAP, Fis et HU, sont apparus comme les acteurs clés dans la promotion des contacts ADN-ADN à longue portée qui se produisent dans tout le chromosome. Il reste à étudier comment les activités d'organisation de l'ADN de Fis et HU qui sont bien comprises à une plus petite échelle (~1 kb) aboutissent à la formation d'interactions ADN-ADN à longue portée. Néanmoins, certaines des interactions ADN médiées par HU nécessitent la présence de naRNA4. naRNA4 participe également à l'établissement de contacts ADN à longue portée. HU catalyse certains des contacts, pas tous, ce qui suggère que l'ARN participe avec d'autres NAP à la formation de contacts ADN. HU semble également agir avec MukB pour favoriser les interactions ADN-ADN à longue portée. Cette hypothèse est fondée sur des observations selon lesquelles l'absence de HU ou de MukB a entraîné une réduction des contacts ADN-ADN. On ne sait pas exactement comment MukB et HU agissent ensemble pour favoriser les interactions ADN-ADN. Il est possible que les deux protéines interagissent physiquement. Alternativement, alors que MukBEF extrude de grandes boucles d'ADN, HU condense et organise ces boucles.
Rôle de la parenté fonctionnelle des gènes
Il existe des rapports selon lesquels les gènes fonctionnellement apparentés d' E. coli sont physiquement ensemble dans l'espace 3D au sein du chromosome, même s'ils sont éloignés par la distance génétique. La proximité spatiale des gènes fonctionnellement apparentés rend non seulement les fonctions biologiques plus compartimentées et efficaces, mais contribuerait également au repliement et à l'organisation spatiale du nucléoïde. Une étude récente utilisant des marqueurs fluorescents pour la détection de loci d'ADN spécifiques a examiné les distances physiques par paires entre les sept opérons d'ARNr qui sont génétiquement séparés les uns des autres (jusqu'à deux millions de pb). Elle a rapporté que tous les opérons, à l'exception de rrn C, étaient à proximité physique. Étonnamment, les études 3C-seq n'ont pas révélé le regroupement physique des opérons rrn , ce qui contredit les résultats de l'étude basée sur la fluorescence. Par conséquent, des recherches plus approfondies sont nécessaires pour résoudre ces observations contradictoires. Dans un autre exemple, GalR forme un réseau d'interaction de sites de liaison GalR qui sont dispersés à travers le chromosome. GalR est un régulateur transcriptionnel du régulon du galactose composé de gènes codant des enzymes pour le transport et le métabolisme du sucre D-galactose. GalR n'existe que dans un ou deux foyers dans les cellules et peut s'auto-assembler en grandes structures ordonnées. Par conséquent, il semble que GalR lié à l'ADN se multimérise pour former des interactions à longue distance.
Forme et structure globales
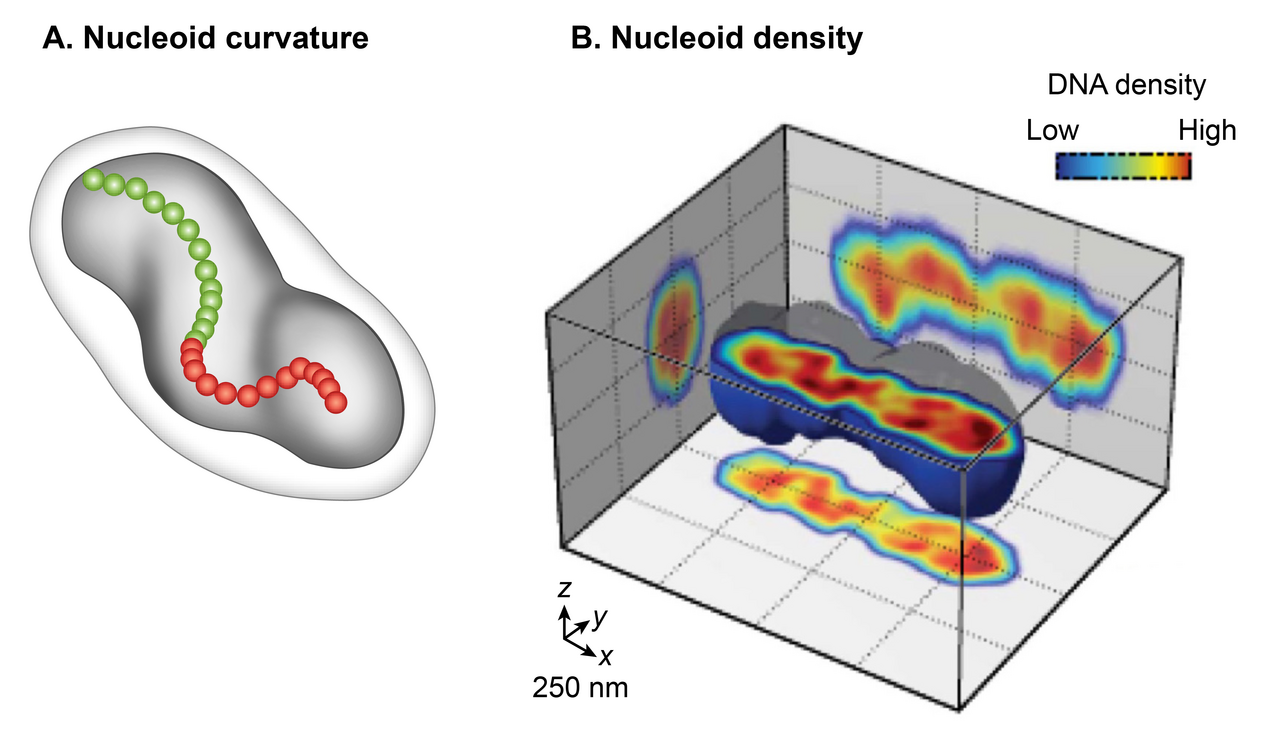
Français La microscopie électronique à transmission (MET) conventionnelle de cellules d'E. coli fixées chimiquement a représenté le nucléoïde comme un organite de forme irrégulière. Cependant, l'imagerie de fluorescence à grand champ de nucléoïdes vivants en 3D a révélé une forme ellipsoïdale discrète. La superposition d'une image en contraste de phase de la cellule et de l'image fluorescente du nucléoïde a montré une juxtaposition étroite uniquement dans la dimension radiale sur toute sa longueur du nucléoïde à la périphérie de la cellule. Cette découverte indique un confinement radial du nucléoïde. Un examen détaillé de l'image de fluorescence 3D après une coupe transversale perpendiculaire à son axe long a en outre révélé deux caractéristiques globales du nucléoïde : la courbure et les régions longitudinales à haute densité. L'examen de la chiralité de la ligne centrale du nucléoïde en reliant le centre d'intensité de chaque coupe transversale a montré que la forme globale du nucléoïde est incurvée. La distribution de l'intensité de fluorescence dans les coupes transversales a révélé une sous-structure de densité, composée de régions ou de faisceaux incurvés à haute densité au niveau du noyau central et de régions à faible densité à la périphérie. L'une des implications du confinement radial est qu'il détermine la forme incurvée du nucléoïde. Selon un modèle, le nucléoïde est forcé de se courber parce qu'il est confiné dans une cellule cylindrique d'E. coli dont le rayon est inférieur à sa longueur pliable (longueur de persistance). Ce modèle a été soutenu par des observations selon lesquelles l'élimination de la paroi cellulaire ou l'inhibition de la synthèse de la paroi cellulaire augmentait le rayon de la cellule et entraînait une augmentation concomitante du rayon hélicoïdal et une diminution du pas hélicoïdal dans le nucléoïde.
Connexions nucléoïde-membrane
Une force d'expansion due aux connexions ADN-membrane semble fonctionner en opposition aux forces de condensation pour maintenir un niveau de condensation optimal du nucléoïde. Des études de fractionnement cellulaire et de microscopie électronique ont d'abord indiqué la possibilité de connexions ADN-membrane. Il existe maintenant plusieurs exemples connus de connexions ADN-membrane. La transertion est un mécanisme de transcription, de traduction et d'insertion simultanées de protéines membranaires naissantes qui forme des contacts ADN-membrane transitoires. Il a été démontré que la transertion de deux protéines membranaires LacY et TetA provoque le repositionnement des loci chromosomiques vers la membrane. Un autre mécanisme de connexions nucléoïde-membrane se fait par un contact direct entre les régulateurs de transcription ancrés à la membrane et leurs sites cibles dans le chromosome. Un exemple d'un tel régulateur de transcription dans E. coli est CadC. CadC contient un domaine sensoriel périplasmique et un domaine de liaison à l'ADN cytoplasmique. La détection d'un environnement acide par son domaine sensoriel périplasmique stimule l'activité de liaison à l'ADN de CadC, qui active ensuite la transcription de ses gènes cibles. La localisation membranaire des gènes régulés par un régulateur de transcription ancré à la membrane n'a pas encore été démontrée. Néanmoins, l'activation des gènes cibles dans le chromosome par ces régulateurs devrait entraîner un contact nucléoïde-membrane, bien qu'il s'agisse d'un contact dynamique. Outre ces exemples, le chromosome est également spécifiquement ancré à la membrane cellulaire par l'interaction protéine-protéine entre les protéines liées à l'ADN, par exemple SlmA et MatP, et le divisome . Étant donné que les gènes codant pour les protéines membranaires sont répartis dans tout le génome, les contacts dynamiques ADN-membrane par transfert peuvent agir comme une force d'expansion nucléoïde. Cette force d'expansion fonctionnerait en opposition aux forces de condensation pour maintenir un niveau de condensation optimal. La formation de nucléoïdes hautement condensés lors de l'exposition des cellules d'E. coli au chloramphénicol, qui bloque la traduction, fournit un support à la force d'expansion des contacts ADN-membrane transitoires formés par la transertion. La forme ronde des nucléoïdes trop condensés après traitement au chloramphénicol suggère également un rôle des contacts ADN-membrane médiés par la transertion dans la définition de la forme ellipsoïdale du nucléoïde.
Visualisation
Le nucléoïde peut être clairement visualisé sur une micrographie électronique à très fort grossissement , où, bien que son apparence puisse différer, il est clairement visible contre le cytosol . Parfois, même des brins de ce que l'on pense être de l'ADN sont visibles. En colorant avec la coloration de Feulgen , qui colore spécifiquement l'ADN, le nucléoïde peut également être vu au microscope optique . Les colorants intercalaires de l'ADN DAPI et bromure d'éthidium sont largement utilisés pour la microscopie à fluorescence des nucléoïdes. Il a une forme irrégulière et se trouve dans les cellules procaryotes.
Dommages et réparation de l'ADN
Des changements dans la structure du nucléoïde des bactéries et des archées sont observés après exposition à des conditions endommageant l'ADN. Les nucléoïdes des bactéries Bacillus subtilis et Escherichia coli deviennent tous deux significativement plus compacts après une irradiation UV. La formation de la structure compacte chez E. coli nécessite l'activation de RecA par des interactions RecA-ADN spécifiques. La protéine RecA joue un rôle clé dans la réparation par recombinaison homologue des dommages à l'ADN.
Comme pour B. subtilis et E. coli ci-dessus, l'exposition de l'archée Haloferax volcanii à des stress qui endommagent l'ADN provoque une compaction et une réorganisation du nucléoïde. La compaction dépend du complexe protéique Mre11-Rad50 qui catalyse une étape précoce de la réparation par recombinaison homologue des cassures double brin de l'ADN. Il a été suggéré que la compaction du nucléoïde fait partie d'une réponse aux dommages de l'ADN qui accélère la récupération cellulaire en aidant les protéines de réparation de l'ADN à localiser des cibles et en facilitant la recherche de séquences d'ADN intactes lors de la recombinaison homologue.